
Review Article
Austin J Anal Pharm Chem. 2014;1(3): 1015.
Small-Molecule Inhibitors of the Mcl-1 Oncoprotein
Lijia Chen1, Maryanna E. Lanning1 and Steven Fletcher1,2*
1Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, MD, USA
2University of Maryland Greenebaum Cancer Center, Baltimore, MD, USA
*Corresponding author: :Steven Fletcher, Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, University of Maryland Greenebaum Cancer Center, USA
Received: September 18, 2014; Accepted: October 05, 2014; Published: October 10, 2014
Abstract
Mcl-1, an anti-apoptotic member of the Bcl-2 protein family, has emerged as an especially attractive target for the development of next generation antineoplastics owing to its direct involvement in tumor genesis as well as its role in the resistance of many cancers to current anti-cancer therapies. The efficacy of Navitoclax against a range of different cancer models that are dependent on Bcl-xL and Bcl-2, two relatives of Mcl-1, indicates that the inhibition of Mcl-1 with small-molecules might also be a viable strategy to kill cancer cells by inducing apoptosis. Herein, we provide a comprehensive review of the most potent, Mcl-1 selective small-molecule inhibitors reported in the literature to date.
Introduction
The Bcl-2 (B cell lymphoma 2) family of proteins, whose anti-apoptotic members include Bcl-2, Bcl-xL, Bcl-w, A1 and Mcl-1 (myeloid cell leukemia 1), are critical regulators of apoptosis, or programmed cell death [1]. A hallmark of cancer is the evasion of apoptosis, and many cancers are resistant to apoptosis due to overexpression of one or more of the Bcl-2 family members [2-4]. ABT-263 (Navitoclax), the clinical analogue of ABT-737, is a potent, dual inhibitor of Bcl-xL and Bcl-2, and is an effective killer of cancer cells over expressing these proteins [5]. However, Navitoclax exhibits limited activity against Mcl-1, such that cancer cells overexpressing Mcl-1 or both Bcl-xL and Mcl-1 are resistant to the drug [6]. Over expression of the Mcl-1 protein and/or amplification of the Mcl-1 gene have been linked to a variety of human cancers, including lung, breast, pancreatic, cervical and ovarian cancers, as well as leukemia and lymphoma [7-15]. Mcl-1 overexpression is directly responsible for the emerging resistance of various FDA-approved anti-cancer therapies, including vincristine [16], Taxol [16], gemcitabine [17] and cisplatin [18]. Importantly, down regulation of Mcl-1 by RNAi decreases tumor-igenicity in mouse xenograft models [17]. Taken together, these observations suggest that the inhibition of Mcl-1 by small-molecules might provide an alternative strategy to kill cancer cells by inducing apoptosis. With this in mind, the focus of this mini-review is on selective small-molecule inhibitors of the Mcl-1 protein; small-molecule inhibitors of Bcl-2, Bcl-xL and dual Bcl-xL/Mcl-1 inhibitors are reviewed elsewhere [19].
Like the other anti-apoptotic Bcl-2 proteins, there is a hydrophobic grove on the surface of Mcl-1 that engages the BH3 “death” domains of the pro-apoptotic Bcl-2 proteins, such as Bim, Bak and Bad. The BH3 domain is an amphipathic α-helix whose hydrophobic face recognizes four hydrophobic sub-pockets, p1, p2, p3 and p4, in the BH3-binding groove on Mcl-1, while a critical Asp on the polar face of the BH3 helix binds Arg263 of Mcl-1[1,20]. Figure 1A shows the crystal structure of the Bim-BH3–Mcl-1 complex, and Figure 1B is the BH3-binding groove on the surface of Mcl-1 with key binding regions and residues highlighted. Through this protein–protein interaction (PPI), Mcl-1 (and the other anti-apoptotic Bcl-2 proteins) “neutralizes” the cell-killing function of the pro-apoptotic Bcl-2 proteins. In this way, overexpression of Mcl-1 leads to the evasion of apoptosis and, hence, cancer. Small-molecules that can block this PPI through binding the hydrophobic groove on Mcl-1 are expected to counter this neutralization and allow the pro-apoptotic proteins to kill the cancer cell through restoring apoptosis. Dual Bcl-xL/ Mcl-1 inhibitors have been reported for about ten years, and these compounds are predicted and/or known to bind in some or all of these sub-pockets [19]. Although Bcl-xL-selective inhibitors, such as ABT- 737, have been identified, there have been limited advances towards the development of Mcl-1-selective inhibitors. In the last 18 months, however, some exciting advances have been made. This mini-review reports the recent progress in the discovery of small-molecules that selectively inhibit the Mcl-1 oncoprotein.
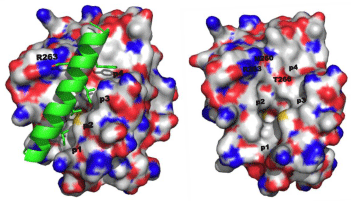
Figure 1: A. (left) Co-crystal structure of the Mcl-1–Bim-BH3 complex (PDB ID: 2PQK); B. (right) The BH3-binding crevice on Mcl-1 highlighting the four sub-pockets p1, p2, p3 and p4, along with key residues involved in the PPI and/or targeted by small molecules in the below sections.
Merged Compounds: 2-Carboxylic Acid-Substituted Benzofurans, Benzothiophenes and Indoles
In seminal work, Fesik’s group at Vanderbuilt University was the first to rationally design selective, small-molecule inhibitors of Mcl-1[20]. In a similar vein to the discovery of the Bcl-xLinhibitor ABT-737 in which he was also involved whilst at Abbott, Fesik and co-workers used NMR-based screening of a large fragment library followed by structure-based design to generate potent Mcl- 1 inhibitors. Fragments were broadly separated into two classes. Class I mostly constituted carboxylic acids attached to a 6, 5-fused hetero cycle, for example 1(Figure 2), which bound Mcl-1 with a Ki of 131 μM, according to a fluorescence polarization competition assay (FPCA), whilst class II was populated by hydrophobic aromatics tethered to a polar functional group, most typically a carboxylic acid as in 2 (Ki = 60 μM). NOESY NMR-guided molecular modeling informed Fesik’s group that the class I compounds were binding near Arg263 , which was likely engaging in salt bridges with the carboxylic acids of the fragments. Also, it appeared the class II compounds were binding deep into the hydrophobic p2 pocket and, when present, the carboxylic acid was located at the surface of this pocket near Arg263. The authors observed that the binding of class I and class II compounds to Mcl-1 was mutually exclusive, suggesting that the carboxylic acid in each class was binding the same residue, possibly Arg263.
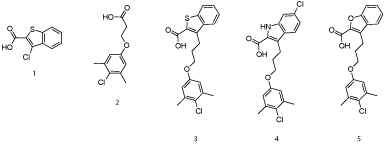
Figure 2: Fesik’s fragment-based drug discovery approach identified potent inhibitors of Mcl-1[20].
With all of this data in hand, small-molecule fragments from each class were then merged together to deliver Mcl-1 inhibitors with affinities that were greatly improved over either fragment alone. Merged compound 3 exhibited a Ki of 320 nM for Mcl-1, representing an almost 200-fold improvement in affinity over the strongest binding fragment 2, and around a 40-fold selectivity over Bcl-xL. A subsequent structure–activity relationship (SAR) campaign improved the affinity to a Ki of 55 nM for compound 4, which was more than 270-fold selective for Mcl-1 over Bcl-xL. Replacement of the heterocyclic S or NH with an isosteric O, such as in 5, resulted in reduced binding affinity of up to 10-fold. A co-crystal structure of 3 with Mcl-1 revealed that the 4-chloro-3, 5-dimethylphenyl group binds deep in the p2 pocket, whilst the carboxylic acid interacts with Arg263 (Figure 3; PDB ID: 4HW3). Overlaying this crystal structure with the binding mode of a 16-mer BH3 peptide derived from Mcl-1 (PDB ID: 4HW4) demonstrated that their small-molecules do not make contacts normally made by Ile210, Val216, Asp218 and Val220 of the BH3 peptide, hinting at possible strategies to enhance the affinity of their Mcl-1 inhibitors further still. Importantly, the observed selectivity of their ligands may be attributed to the occupation of the p2 pocket, which, although present in Bcl-xL, is larger and deeper in Mcl-1. In addition, the co-crystal structure of 3 with Mcl-1 disclosed that the p2 pocket opens up somewhat to accommodate the 4-chloro-3, 5-dimethylphenyl moiety indicating some plasticity in this region. At the time of writing this review, no cell activity has been reported on these Mcl-1 inhibitors. Regardless, following in the footsteps of the discovery of ABT-737, Fesik’s work has yet again paved the way for the development of potent and selective inhibitors of another “undruggable” PPI.
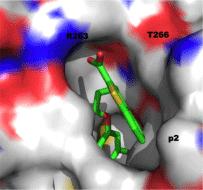
Figure 3: Co-crystal structure of benzothiophene 3 with Mcl-1 (PDB ID: 4HW3).
MIM1, A Substituted Pyrogallol
Walenksy and co-workers used a fluorescently-labeled, hydrocarbon-stapled Mcl-1 BH3 helix as the probe to screen a large library of compounds, which led to the discovery of MIM1 (Mcl-1 Inhibitor Molecule 1, 6 (Figure 4)) that binds in the BH3-binding groove on the surface of Mcl-1[21]. The screening process began with 71,296 small molecules, which were whittled down to 64 potent and selective Mcl-1 inhibitors, many of which could be organized into a few discrete structural classes. Further refinement was afforded by dose response studies in FPCAs with Mcl-1 and Bcl-xL, and cytotoxicity assays, which ultimately led to the identification of MIM1. Not surprisingly, since this group and variants feature in several dual Bcl-xL/Mcl-1 inhibitors, the pyrogallol (benzene-1,2,3,-triol) moiety was required for activity. However, MIM1 is the first Mcl-1-selective inhibitor that bears this motif: MIM1 disrupted the complex between Mcl-1 and a fluorescently labeled Bid BH3 peptide (FITC-Bid BH3) with an IC50 value of 4.8 μM, whilst MIM1 demonstrated no capacity to disrupt the FITC-Bid BH3–Bcl-xL complex (IC50> 50 μM).
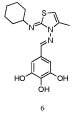
Figure 4: “MIM1” [21].
NMR structural studies of MIM1 with 15N-Mcl-1 revealed that the small-molecule binds in the canonical BH3 binding groove. Subsequent computational modeling generated a putative binding mode of MIM1. The cyclohexyl group was predicted to make hydrophobic contacts near the p3 pocket, while the thiazolyl core and its methyl substituent probe deeply into the p2 pocket. In comparison with other Mcl-1-selective inhibitors, this latter interaction may be a source of MIM1’s selectivity and further structural modification here might enhance selectivity further still. The pyrogallol motif forms hydrogen bonds with Asp256 and Arg263, which are residues that are engaged by the hydrophilic face of the amphipathic BH3 α-helices. Finally, MIM1 was shown to selectively inhibit Mcl-1- based suppression of pro-apoptotic Bax activation through freeing up the BH3-only protein tBID, and selectively induced cell death in an Mcl-1-dependent leukemia cell line.
3-Substituted-N-(4-hydroxynapthalen-1-yl) Aryl Sulfonamides
Similarly to Walensky’s approach, Nikolovska-Coleska’s laboratory also applied a high throughput screening strategy to identify Mcl-1 inhibitors [22]. After screening a small-molecule library of over 50,000 compounds by a FPCA, the authors discovered compound 7 that bound Mcl-1 with a Ki of 1.55 μM, which is around the same affinity as MIM1. Computational modeling predicted that the naphthalene ring binds in the p3 pocket, whilst the thiophene binds in the p2 pocket. The carboxylic acid forms a network of hydrogen bonds with Arg263 and Asn260, and the phenolic hydroxyl group binds His224. HSQC NMR spectroscopy with 15N-Mcl-1 confirmed that 7 (Figure 5) bound the hydrophobic groove on the surface of Mcl-1 and, therefore, was functioning as a BH3 mimetic. With this data in hand, Nikolovska-Coleska and co-workers embarked on a structure-based design approach to optimize their hit compound. A library of around 50 derivatives was prepared, of which the most potent member 8 bound Mcl-1 with a Ki of 180 nM. Compound 8 was selective for Mcl-1 over the other anti-apoptotic Bcl-2 proteins, most notably almost 60-fold selective over Bcl-xL.
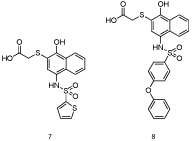
Figure 5: 3-Substituted-N-(4-hydroxynapthalen-1-yl) aryl sulfonamides [22].
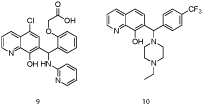
Figure 6: 7-Substituted 8-hydroxyquinolines [24].
A subset of compounds was analyzed for their abilities to inhibit cell proliferation in the leukemia cell lines HL-60, MV4-11 and K-562. HL-60 and MV4-11 cells are sensitive to Mcl-1 inhibition, whereas K-562 cells are not. Pleasingly, low micromolar IC50 values were observed, and, in many cases, several-fold selectivities were reported for HL-60 and/or MV4-11 cells over K-562 cells. Our own research group has independently prepared some compounds that are similar to the naphthalene derivatives reported by Nikolovska- Coleska’s group. Although we, too, see potent inhibition of Mcl-1, we do not observe any appreciable cellular activity [23]. It is well known that carboxylic acids can inhibit cell penetration due to their charged state at physiological pH. Although not discussed in their paper, we speculate that Nikolovska-Coleska’s Mcl-1 inhibitors might reversibly from a neutral, six-membered lactone through cyclization of the carboxylic acid and the phenol hydroxyl group, allowing passage across the cell membrane. Once inside the cell, hydrolysis back to the hydroxy acid regenerates the active inhibitor. Our inhibitors are incapable of forming such neutral, internal prodrugs, which may explain the differences in cell activities. Finally, it was also demonstrated that compound 8 selectively caused cell death in Mcl-1-overexpressing lymphoma cells through a Bak/Bax-dependent mechanism, and which correlated with activation of caspase-3 and induction of apoptosis. Altogether, this biochemical data point towards 8 functioning as an Mcl-1 selective BH3 mimetic within cells.
8-Hydroxyquinolines
In a collaborative effort between Eutropics Pharmaceuticals and researchers at The Scripps Research Institute, a high throughput screen of the NIH Molecular Libraries and Small Molecule Repository (MLSMR) led to the discovery of a selective Mcl-1 inhibitor (IC50 = 2.4 μM) based on an 8-hydroxyquinoline-derivative scaffold (9) [24]. Towards acquiring more “drug-like” characteristics, the carboxylic acid, which might otherwise impede cell entry, was removed, as was the 4-chloro group since this typically enhances lipophilicity and reduces solubility. Gratifyingly, Mcl-1 affinity and selectivity were retained. A subsequent round of SAR analysis revealed that the 8-hydroxyl group and the quinoline nitrogen were essential. Next, modification of the phenyl and amino-pyridine resulted in compound 10, which exhibited excellent affinity for Mcl-1 (IC50 = 0.31 μM) and selectivity over Bcl-xL (IC50> 40 μM). Molecular modeling with the R-enantiomer of 10 suggested that the 8-hydroxyquinoline moiety engages in a hydrogen bond with Asn260, which orients the N-ethylpiperazine and para-CF3-phenyl groups for delivery into the p2 and p4 pockets, respectively.
In cell-based assays, 8-hydroxyquinoline derivative 10 was anti proliferative in the low micromolar range (EC50 s = 0.3 – 15 μM) in a diverse panel of human-derived cancer cell lines, including the lymphoma cell line Mcl1-1780. Subsequently, BH3 profiling was enlisted to determine the dependency of these cell lines on the anti-apoptotic Bcl-2 proteins for survival; EC50s and profiling demonstrated very good correlation (R2 = 0.81), suggesting the growth inhibition of 10 may be closely associated to inhibition of the Bcl-2 proteins. Compound 10 was then shown to induce cytochrome c release and nuclear apoptosis, both of which are consistent with the inhibition of the anti-apoptotic Bcl-2 proteins. Moreover, the extent of apoptotic induction of 10 was greater in the Mcl-1-overexpressing cell line Mcl1-1780, reflecting the selectivity observed for Mcl-1 in vitro.
2-(Arylsulfonamido) Benzoates and 2-Hydroxybenzoates (Salicylates)
Researchers at AbbVie employed fragment-based methods to identify two novel series of Mcl-1 inhibitors, one based on a 2-(arylsulfonamido) benzoate scaffold and the other on a salicylic acid motif [25]. Recombinant, human Mcl-1, in which the terminal methyl groups of valine, methionine, leucine and isoleucine were 13C-labeled was utilized in a 13C-HSQC NMR-based screen of a library of 17,000 molecules. The top “hits” by NMR were further analyzed by FPCA, which, along with consideration of ligand efficiency (LE) and synthetic tractability, propelled aryl sulfonamide and salicylic acid derivatives to the top of the list. An NMR-derived model of the binding mode of aryl sulfonamide 11 (IC50 = 5 μM; Figure 7) suggested the vinyl group was oriented towards the p2 pocket normally occupied by Leu62 of the Bim-BH3 peptide. A focused library in which the vinyl group was replaced with various aryl groups afforded more than a ten-fold improvement in Mcl-1 inhibitory activity, as embodied in 12 with an IC50 of 0.4 μM. Subsequent modification of the solvent-exposed sulfonamide moiety delivered even more potent inhibitors with the best of the series carrying a pyrazole appendage (13: IC50 = 30 nM). A co-crystal structure of 14 (IC50 = 0.5 μM; Figure 8; PDB ID: 4OQ5) revealed that the distal phenyl ring of the biphenyl ether moiety is projected into the p1 pocket, the naphthyl group into the p2 pocket and the carboxylic acid binds Arg263, akin to Asp67 of the Bim-BH3 peptide. It is particularly noteworthy that the p2 pocket opens up somewhat, allowing much deeper penetration of the naphthyl group than Leu62 of Bim-BH3. A similar finding was observed by Fesik and colleagues, and it has been proposed that this may be a source of Mcl- 1 selectivity by synthetic ligands.
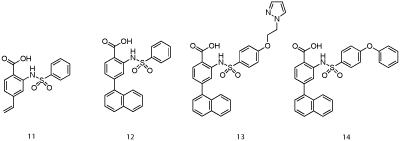
Figure 7: AbbVie’s aryl sulfonamide-based Mcl-1 inhibitors [24].
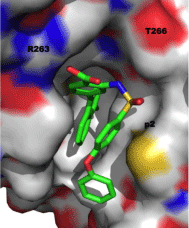
Figure 8: Co-crystal structure of aryl sulfonamide 14 with Mcl-1 (PDB ID: 4OQ5).
Computational modeling of salicylic acid derivative 15 (IC50 = 160 μM; Figure 9) indicated structural elaboration at the 5-position might enhance binding affinity through engaging in additional hydrophobic contacts with the p1 pocket (the aryl ring of 15 was close to the p2 pocket) [25]. Pleasingly, a small library of biphenyls derived from 15 demonstrated improved activities, with submicromolar IC50 values observed. Their most potent member of this class was 16 with an IC50 of 0.57 μM. A co-crystal structure of salicylate 17 (Figure 10; PDB ID: 4OQ6) confirmed the predicted binding mode, including the salt bridge interaction between the carboxylic acid and Arg263 (which was actually a constraint in the molecular modeling experiments). In summary, utilizing a combination of NMR-directed, fragment-based drug discovery and structural biology, Petros and co-workers at AbbVie have successfully enhanced Mcl-1 inhibitory activity of initial hits by almost 300-fold. Since the naphthyl moiety of aryl sulfonamide 13 probes deeply into the p2 pocket of Mcl-1, it will be interesting to determine if this imparts selectivity over Bcl-xL, as appears to be the case with Fesik’s Mcl-1 inhibitors. Finally, it is unclear if either the salicylic acid and aryl sulfonamide derivatives reported within this paper exhibit cellular activity, as this was not discussed. If cell activity is lacking, then ester prodrugs and carboxylic acid bioisosteres might address this inadequacy and allow these exquisitely simple PPI inhibitors to be advanced beyond in vitro studies.
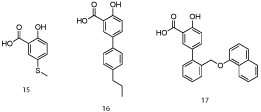
Figure 9: AbbVie’s 5-phenylsalicylate-based Mcl-1 inhibitors [25].

Figure 10: Co-crystal structure of salicylate 17 with Mcl-1 (PDB ID: 4OQ6).

Figure 11: Deconstruction of dual Mcl-1/Bcl-2 inhibitor 18 followed by rebuilding into Mcl-1 selective inhibitor 20 [26].
8-Oxo-3-thiomorpholino-8H-acenaphtho [1, 2-b] pyrrole-9-carbonitrile and Fragments
Zhang and colleagues used a fragment-based approach to convert their previously-reported dual Mcl-1/Bcl-2 inhibitor 8-oxo- 3-thiomorpholino-8H-acenaphtho-[1, 2-b]pyrrole-9-carbonitrile (18: Kd = 58 nM (Mcl-1), 310 nM (Bcl-2)) into a more “drug-like” and Mcl-1 selective inhibitor [26]. Compound 18 was dissected into several fragments of which cyanoacetamide (19) exhibited good binding affinity to Mcl-1 (Kd = 13.5 μM) and the highest LE. To gather information on the likely binding mode of 19, the authors prepared an R263A mutant of Mcl-1 since this arginine plays a significant role in the recognition of the Bim-BH3 helix through engaging D67 of Bim in a salt bridge. Fragment 19 demonstrated no appreciable affinity (Kd> 1000μM) to the mutant Mcl-1 protein, indicating it binds R263, possibly through a hydrogen bond in which the carbonyl of 19 serves as the hydrogen bond acceptor. Molecular modeling studies suggested that functionalization of the CH2 and NH2 groups of 19 might allow occupation of the p2 and p4 pockets, respectively. Accordingly, hydrophobic moieties were added to these functional groups.
Resulting second generation compound 20 demonstrated an improvement in affinity for Mcl-1 (Kd = 0.16 μM) of almost two orders of magnitude over fragment 19. HSQC NMR spectroscopy with 15N-Mcl-1 revealed chemical shift perturbations of side chains located primarily in the p2 and p4 pockets, as well as that of R263, providing experimental validation of the authors’ proposed binding mode. A FPCA indicated that 20 exhibited no affinity for Bcl-2, and, therefore, that dual Mcl-1/Bcl-2 inhibitor 18 had been transformed into an Mcl-1 selective inhibitor. Compound 20 selectively induced apoptosis in the Mcl-1-dependent cell line NCI-H23 with an IC50 of 0.38 μM over cell lines that are dependent on Bcl-2, mirroring the in vitro data. Finally, 20 demonstrated improved aqueous solubility over parent 18. While 20 does not appear to be generally toxic, its further drug development should be heeded with caution since it bears Michael acceptor acrylonitrile and acrylamide moieties.
2-Hydroxynicotinonitrile and Derivatives
In an alternate dissection of 18, Zhang et al. identified a fragment related to cyanoacetamide (19), specifically 2-hydroxynicotinonitrile (21) [27]. Although less potent with a lower LE than 19 (Ki = 113 μM, LE = 0.60), fragment 21 represented an additional starting point from which to develop Mcl-1 inhibitors. The incorporation of a methoxy group at the 4-position of the pyridine ring resulted in a loss of binding affinity, so further modifications were focused on the 5-position. A small library of compounds was subsequently prepared that included aryl and thioaryl substituent’s at this site, all of which led to enhanced affinity for Mcl-1 over the parent compound 21. Notably, the Ki was improved by more than two orders of magnitude for derivative 22 (Ki = 0.86 μM). Further enhancements in affinity for Mcl-1 were obtained by conversion of the nitrile group to N-functionalized carboxamides, possibly through gaining access to the p4 pocket (IC50s (ELISA) = 0.890 μM (22), 0.054 μM (23)). Isothermal titration calorimetry (ITC) provided evidence that 23 was selective for Mcl-1 over Bcl-2. Furthermore, cell assays revealed that 23 selectively induced apoptosis in the Mcl-1 dependent K562 cancer cell line (IC50 = 0.84 μM) over the Bcl-2 dependent RS4; 11 cancer cell line (IC50 = 24.5 μM). Taken together, Zhang and colleagueshave identified a second, chemically diverse scaffold that is ready for further optimization.

Figure 12: Mcl-1 inhibitors based on 2-hydroxynicotinonitile and 2-hydroxynicotinamide cores [27].
N-Benzyl Piperazines
Employing a computational algorithm, Ding and co-workers designed four series of benzyl piperazine derivatives as putative inhibitors of the Bcl-2 proteins, which are somewhat reminiscent of ABT-737 [28]. Of 81 compounds synthesized, 22 exhibited binding affinities in the low micromolar range to at least one of Mcl-1, Bcl-xL and Bcl-2. Although not a primary goal of their research, a few compounds were found to be selective for Mcl-1 with no appreciable affinity to the other two Bcl-2 proteins investigated. In particular, 24, comprised of a benzyl piperazine linked to tri-methylated pyrogallol motif, bound Mcl-1 with a Ki of 0.18 μM. Molecular modeling was used to attempt to explain the relative selectivities of some of the compounds, although HSQC NMR spectroscopy with 15N-Mcl-1 or co-crystal studies should be conducted to obtain experimental data that will provide information on binding modes and, thereby, assist in the design of more potent and more selective, second-generation Mcl-1 inhibitors.

Figure 13: An N-benzyl piperazine coupled to a tri-methylated pyrogallol motif [28].
Outlook
The overexpression of Mcl-1 has emerged as a critical factor in the resistance to apoptosis of a wide range of cancers, and, therefore, its inhibition has become a highly sought after goal. Although Bcl-xL-selective and dual Bcl-xL/Mcl-1 inhibitors have been known for around ten years, Mcl-1-selective inhibitors remained elusive until 2012. The recent co-crystal structures of small-molecules bound to Mcl-1 reported by Fesik, and then by AbbVie, reveal insights into features that might be exploitable to accomplish Mcl-1 selectivity and potency with new ligands as well as in the further optimization of existing ligands. For example, the p2 pocket is larger and deeper in Mcl-1 than in Bcl-xL, and also appears to open up in the presence of hydrophobic ligands. Additionally, many of the Mcl-1 selective ligands reported herein do not bind the p4 pocket, which is open to the solvent in Mcl-1 but in Bcl-xL is more enclosed. In order to craft Mcl-1 selective inhibitors, we, therefore, propose the following criteria. The p2 pocket should be targeted with hydrophobic moieties that are flexibly linked to the scaffold to allow optimal occupation of the plastic p2 pocket. Hydrogen-bond donor and acceptor groups should be included to bind Arg263 and/or Asn260, which may be negatively-charged or uncharged. Hydrophobic groups may also be incorporated to bind the p1 and p3 pockets to furnish additional binding affinity, although it may be advantageous to avoid targeting the p4 pocket as this may erode Mcl-1 selectivity through better targeting of Bcl-xL.
References
- Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9: 47-59.
- Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
- Hanahan D, Weinberg RA . The hallmarks of cancer. Cell. 2000; 100: 57-70.
- Fulda S, Debatin KM . Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006; 25: 4798-4811.
- Tse C1, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF . ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008; 68: 3421-3428.
- Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al . Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007; 67: 1176-1183.
- Warr MR, Shore GC . Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr Mol Med. 2008; 8: 138-147.
- Backus HH, van Riel JM, van Groeningen CJ, Vos W, Dukers DF, Bloemena E, et al . Rb, mcl-1 and p53 expression correlate with clinical outcome in patients with liver metastases from colorectal cancer. Ann Oncol. 2001; 12: 779-785.
- Song L, Coppola D, Livingston S, Cress D, Haura EB . Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005; 4: 267-276.
- Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, et al . Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007; 67: 4564-4571.
- Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, Fujimoto K, et al . Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology. 1999; 56: 73-82.
- Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F . The expression of Mcl-1 in human cervical cancer and its clinical significance. Med Oncol. 2012; 29: 1985-1991.
- BrotinE, Meryet-Figuiére M, Simonin K, Duval RE, Villedieu M, Leroy-Dudal J, et al. Bcl-XL and MCL-1 constitute pertinent targets in ovarian carcinoma and their concomitant inhibition is sufficient to induce apoptosis. Int. J. Cancer 2010; 126: 885–895.
- Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al . Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012; 26: 120-125.
- Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, et al . MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia. 2013; 27: 1381-1390.
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471: 110–114.
- Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, et al. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008; 62: 1055-1064.
- Michels J, Obrist F2, Vitale I3, Lissa D1, Garcia P1, Behnam-Motlagh P4, et al . MCL-1 dependency of cisplatin-resistant cancer cells. Biochem Pharmacol. 2014; .
- Vogler M, Dinsdale D, Dyer MJ, Cohen GM . Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009; 16: 360-367.
- Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, et al . Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013; 56: 15-30.
- Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, et al . A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol. 2012; 19: 1175-1186.
- Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, et al . 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J Med Chem. 2014; 57: 4111-4133.
- Lanning ME, Fletcher S, et al. Unpublished results.
- Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, et al . Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg Med Chem. 2013; 21: 6642-6649.
- Petros AM, Swann SL2, Song D2, Swinger K2, Park C2, Zhang H2, et al . Fragment-based discovery of potent inhibitors of the anti-apoptotic MCL-1 protein. Bioorg Med Chem Lett. 2014; 24: 1484-1488.
- Zhang Z, Song T, Li X, Wu Z, Feng Y, Xie F, et al . Novel soluble myeloid cell leukemia sequence 1 (Mcl-1) inhibitor (E,E)-2-(benzylaminocarbonyl)-3-styrylacrylonitrile (4g) developed using a fragment-based approach. Eur J Med Chem. 2013; 59: 141-149.
- Zhang Z, Liu C, Li X, Song T, Wu Z, Liang X, et al . Fragment-based design, synthesis, and biological evaluation of N-substituted-5-(4-isopropylthiophenol)-2-hydroxynicotinamide derivatives as novel Mcl-1 inhibitors. Eur J Med Chem. 2013; 60: 410-420.
- Ding X, Li Y, Lv L, Zhou M, Han L, Zhang Z, et al . De novo design, synthesis and evaluation of benzylpiperazine derivatives as highly selective binders of Mcl-1. ChemMedChem. 2013; 8: 1986-2014.