
Research Article
Austin J Anal Pharm Chem. 2014;1(5): 1022.
Composite Sodium Alginate and Chitosan Based Wafers for Buccal Delivery of Macromolecules
Boateng JS1* and Areago D1
1Department of Pharmaceutical, Chemical and Environmental Sciences, Faculty of Engineering and Science, University of Greenwich, UK
*Corresponding author: :Boateng JS, Department of Pharmaceutical, Chemical and Environmental Sciences, Faculty of Engineering and Science, University of Greenwich, UK.
Received: October 29, 2014; Accepted: November 06, 2014; Published: November 07, 2014
Abstract
The objective of this study was to develop a composite buccal drug delivery wafer for protein drug delivery. The polymeric vehicle used in this study combined chitosan and sodium alginate with bovine serum albumin (BSA) as a model drug. The wafers were obtained by freeze-drying gels of the polymers in well plates. Prior to the lyophilisation process, differential scanning calorimetry was performed to establish a suitable freeze-drying cycle. Preliminary characterization experiments were undertaken to select the optimum composite gel containing sodium alginate and chitosan in a 4:1 ratio respectively for drug loading. A second series of characterisation tests were performed on the drug-loaded wafers prepared from gels containing 0.25 and 0.5 % w/w of BSA. The formulations were functionally characterised for swelling, mucoadhesive and drug dissolution properties. The morphology and crystallinity were investigated using a scanning electron microscope and X-ray diffractometer respectively. The results from drug dissolution studies over a two-hour period showed 66% and 31% cumulative drug release for the wafers obtained from gels containing 0.25 and 0.50 % w/w BSA respectively. These results show the feasibility of developing a sustained delivery system for macromolecules by combining chitosan and sodium alginate.
Keywords: Chitosan; Sodium alginate; Bovine serum albumin; Buccal delivery; Protein delivery
Abbreviations
BSA (Bovine serum albumin); Ch (Chitosan); DSC (Differential scanning calorimetry); PAF (Peak adhesive force); PBS (Phosphate buffered saline); SA (Sodium alginate); SEM (Scanning electron microscopy); XRD (X-ray diffraction).
Introduction
Mucosal drug delivery has gained interest for drug delivery involving the use of bioadhesive formulations to administer drugs via sites in the body such as buccal [1-4], nasal [5], wound surfaces [6-7] and vaginal [8] mucosa. The intimate contact between the bioadhesive dosage form and mucosa is facilitated by physico-chemical interactions which allow for an improvement of the drug's absorption and subsequent increase in its bioavailability [9]. The drug carrier system, typically a polymer hydrogel, adheres to the mucosa, via a process initiated by hydration and swelling which allows interpenetration between the bioadhesive polymeric chains and the mucin present on the mucosal membrane, resulting in the formation of weak bonds [10]. Mucosal surfaces are targeted as they present highly vascularised networks, which allow therapeutic delivery whilst avoiding pre systemic circulation (first pass metabolism). There are other benefits of mucosal drug delivery systems including increased patient compliance as there is no pain or risk of choking as for injections and tablets respectively. Drug levels remain steady therefore allowing better control; which in turn reduces the risk of toxicity, as well as complete utilization of the drug administered [11].
The lining of the oral cavity comprises the buccal (cheek muscle tissue), palatal, sublingual (floor of the mouth) and gingival areas. The sublingual and buccal areas are more useful for drug delivery as they are more permeable than any other areas of the mouth and represent roughly 60% of the total oral mucosa surface area. Despite having a smaller surface area of 100cm2 compared to the GIT and skin; which are 350,000cm2 and 20,000cm2 respectively, the oral mucosa is an area of significant interest [12]. The buccal mucosa is a highly adaptable area and considered useful for controlled drug release. One of its characteristics is that damaged tissue only requires a short period of time to heal in comparison to other areas [13].
Different active pharmaceutical ingredients with low molecular weights (small molecules) have been administered via the buccal route including analgesics such as fentanyl citrate, ACE inhibitors such as captopril and benzodiazepines such as midazolam [14]. However, the administration of macromolecules such as peptides and proteins are more challenging due to their large size and presence of charge [2, 15] and several approaches have been proposed to overcome these challenges. Ideal systems contain components such as permeation enhancers which can manipulate the site of absorption to enhance partitioning of the drug into the mucosal tissue or modification of the mucosal surface to increase solubility of the drug which increases the concentration gradient to enhance absorption [16]. Other studies have been reported which show that peptide drugs such as insulin can be transported across the mucosa through the use of enzyme inhibitors and bioadhesive polymers such as chitosan [17-20].
Sodium alginate, the salt form of alginic acid is a polysaccharide composed of 1-4 linked α-L-guluronic and β-D-mannuronic acid residues [21] and is widely used in pharmaceutical formulations due to its high bioadhesive nature, aqueous solubility and its good film forming properties [22]. Chitosan is a weak cationic polyaminosaccharide derived from deacetylation of the native polymer chitin [23], present in shellfish and discovered 200 years ago [24]. Chitosan is composed mainly of (1, 4) linked 2-amino-2- deoxy-β-D-glucan [25], and has been developed as a suitable matrix for the controlled release of protein or peptide drugs over the last two decades [26]. It has been reported that "pH sensitive hydrogels such as sodium alginate and chitosan are useful for protein delivery because of the immunogenicity of most synthetic polymers and the requirement for a harsher environment which may denature and inactivate the protein" [27]. Formulation of protein based systems using both alginate and chitosan as matrices can be achieved under relatively mild environments, therefore avoids potential damage to the proteins' native structure.
Formulations combining sodium alginate and chitosan have been reported including particulate systems (e.g. micro and nanoparticles) [28, 29] and tablets [30]. These formulations have mainly been used for small molecules or delivery via sites other than the buccal mucosa. To the best of our knowledge, no study has been reported of freeze-dried wafers combining both sodium alginate and chitosan for protein or peptide delivery via the buccal mucosa. Though similar studies have reported on buccal formulations for protein and peptide delivery, these have involved single polymers, mainly chitosan or its derivatives [4, 11, 15, 18, 19, 20]. Shaikh and co-workers reported that increasing the amount of chitosan in a composite mucoadhesive tablet formulation resulted in more controlled drug (itraconazole) release while an increase in sodium alginate resulted in improved adhesive properties of the tablet [30].
The aim of this study therefore, was to develop a lyophilised wafer as a controlled buccal delivery system for protein drugs using a combination of sodium alginate and chitosan as the polymeric matrices. Bovine serum albumin (BSA) was used as a model drug as it is a naturally occurring protein. The wafers have been characterised using various analytical techniques to evaluate the wafer's functional physical and mechanical properties.
Materials and Methods
Materials
Chitosan (medium molecular weight, 75-85% deacetylated), sodium alginate, bovine serum albumin (BSA), Bradford's reagent, mucin from porcine and gelatine were all purchased from Sigma- Aldrich, Gillingham, UK. Sodium di hydrogen orthophosphate dihydrate; acetic acid, sodium hydroxide pellets were all purchased from Fisher Scientific (UK).
Pre-formulation studies
Initial studies involved development of polymer gels to identify an optimum formulation for drug loading. Sodium alginate (SA) polymer gels were prepared at concentrations of 0.25% 0.5% and 1% w/w in distilled water with magnetic stirring at a temperature of 37°C. Chitosan (Ch) polymer gels were also prepared in the same way at the same concentrations but replacing distilled water with acetic acid solution (0.05 - 1% v/v). To obtain optimum amounts of both polymers in a composite gel (1% w/w solution), different ratios of each polymer were combined to give a total polymer weight of 0.5 g in 50ml of solution as shown in Table 1. The required amount of SA was dissolved in 25ml of distilled water (37°C) with stirring followed by the addition of 25ml of dilute acetic acid solution (0.05 - 1% v/v) after which the required amount of Ch was added. The pH of the final composite gels ranged between 4 and 5. Based on the previous criteria, the gel with SA: Ch ratio 4:1 was chosen as the ideal combination and loaded with two different concentrations of BSA (0.25 and 0.50% w/w). Different quantities (1-5 g) of the above gels were poured into separate wells of 24 well plates prior to freeze-drying to produce wafers with different thickness.
![]()
BLANK GELS
Polymer ratios
SA: Ch
SA
(mg)
Ch
(mg)
1:4
100.5
400.5
2:3
200.8
300.8
3:2
300.7
200.5
4:1
400.7
100.4
DRUG LOADED GELS
BSA concentration
(w/w)
SA
(mg)
Ch
(mg)
BSA
(mg)
0.50
400.2
100.4
250.9
0.25
400.5
100.2
125.3
Table 1: Composition of blank combined gels containing sodium alginate (SA) and chitosan (Ch) in different ratios with total polymer concentration of 1% w/w and drug loaded gels with SA:Ch ratio of 4:1 containing two different concentrations of BSA (0.25% w/w and 0.5% w/w).
Differential scanning calorimetry (DSC)
Prior to freeze-drying, pure polymers and combined gel (SA: Ch 4:1) were analysed using DSC based on a previously reported method [11]. The gels were cooled in 40 μl aluminum pans with pierced lids from 25 to -55°C at a rate of 10°C/min. They were then re-heated back to 25°C at a rate of 20°C/min and the cycle repeated three times.
Freeze-drying
The freeze-drying procedure involved a 3-stage process: freezing, primary drying and secondary drying on an Advantage Plus Freeze- Drying Machine (Biopharma Processing Systems, UK) over a 48 hour period using an automated cycle previously reported by Kianfar and co-workers [3]. The wafers were removed from the machine and immediately placed in sealed plastic envelopes and stored over silica gel at room temperature until required.
Functional physical-mechanical characterisation
The wafers were characterised for functional properties essential for an effective buccal mucosa protein delivery system as described below.
Hardness testing
The resistance of the wafers to compressive deformation ('hardness') was measured with the help of a texture analyzer machine (Stable Microsystems, UK) in compression mode, using a 6mm (P6) cylindrical probe. The test allows assessment of the structural integrity of the wafer as whole by applying a known force across the diameter of the wafer. The experimental procedure employed was modified from a method previously reported [33]. The settings used were pre-test speed 0.5mm/s, test speed 0.5mm/s, post-test speed 1.0mm/s applied force 100g, contact time 5.0sec, trigger type auto and trigger force set a 5.0g. Each wafer obtained by pouring different amounts of gel (1-5g) was assessed at three triangulated points and the average resistance to compression values computed.
Swelling capacity
The swelling capacity allowed for the assessment of the hydration capabilities. A phosphate buffered saline (PBS) solution was prepared by mixing sodium di hydrogen orthophosphate dehydrate and sodium hydroxide in known amounts in distilled water. The pH of the PBS solution was adjusted to 6.8± 0.1 and kept at 37°C ± 1°C to simulate conditions found in the oral cavity. To further mimic the conditions in the mouth 0.5g of mucin was added to obtain a 0.01% mucin solution. Each wafer was placed in a weighing boat and the weight of the wafer was recorded prior to commencing the test. The procedure involved adding 10ml of the PBS based mucin solution to each weighing boat. The swelling behavior was recorded over a period of 20 min at set intervals of 2min, 5 min, 10 min, 15 min and 20 min. At each time point, the samples were blotted to remove excess liquid droplets, weights recorded then a further 10ml of PBS solution added. This was repeated for each wafer prepared above and the swelling capacity calculated using the following equation
Swelling capacity (%) = 100 x (Xi -X) /X............. equation 1
Where Xi is the weight of the hydrated wafer, and X is the initial weight of wafer.
Scanning electron microscopy (SEM)
To the assess the surface topography of the wafers, sliced sections were cut from each wafer and placed on double sided adhesive carbon tape which had been stuck to stainless steel stubs. The samples were sputter coated for 2 minutes at 25 mA and 1 kV with chromium (EmiTECH K575X Sputter Coater, Quorum Technologies Ltd, Kent, UK) and placed in the chamber of a Hitachi SU8030 scanning electron microscope (Hitachi, Krefeld, Germany). SEM images were acquired at an accelerating voltage of 20 kV and working distance of 15 mm and processed with i-scan2000 software [19]. This test was performed for different blank wafers obtained from pouring 1g, 2g and 3g of gel as well as the two drug loaded wafers.
Mucoadhesion
To assess how the wafers would adhere to the buccal area, in vitro mucoadhesive measurements were performed using a texture analyzer (Stable Microsystems, UK). A model buccal mucosal substrate was prepared from gelatin gel (6.67% w/v) [11]. 6.67g of gelatin was dissolved in 100ml of distilled water. This was heated at 60°C ± 1°C for a period of 15 min. 20ml of the gelatin solution was poured into a Petri dish and allowed to solidify. A 2% mucin solution was made by dissolving 0.2g of mucin in 10ml of PBS solution (pH 6.8± 0.1). 500μl of the mucin solution was spread across the solidified gelatin gel surface to simulate the buccal mucosa lining. Double-sided adhesive tape was applied to a P35 adhesive probe and the wafer attached to the other side of the tape. The Petri dish was placed on the instrument platform and the probe was set to just touch the model mucosal surface. The following parameters were applied to the texture analyzer in tensile mode; pre- test speed 0.5mm/s, test speed 0.5mm/s, post-test speed 1.0mm/s applied force 100g, contact time 60.0s, trigger type auto and trigger force set a 5.0g. The maximum force required to detach the wafer from the model mucosal surface, referred to as the peak adhesive force (PAF) was measured. The test was performed on blank wafers obtained from pouring 2 and 3 g of gel as well as the drug loaded wafers (n = 4).
X-ray diffraction (XRD)
This test was performed to investigate the physical form (crystalline or amorphous) of the blank and drug loaded wafers using an X-ray diffractometer (Bruker, Coventry, UK) in transmission mode. For the pure polymers, a small amount was spread onto a clear film after which it was inserted into a sample holder. In the case of the wafers however, the samples were compressed from their original width of 2-3mm to 0.5mm using clean glass slides, after which they were placed in the sample holder. Diffractograms were acquired for each sample over a 2 theta range of 3 - 55°.
In vitro drug dissolution studies
This test was performed to assess the drug releasing profile of the BSA from the two drug loaded formulations. Prior to dissolution studies, calibration solutions (0.01 - 0.05 mg/ml) of BSA were used to plot a calibration curve using Bradford assay. 1ml of each solution was placed in a cuvette followed by the addition of 2ml of Bradford's reagent. The absorbance was measured at 595nm for each dilution. This was possible as the binding of BSA to the dye present in the reagent results in a shift from the reddish-brown colour to a deep blue colour. The dissolution study required the use of the Franz diffusion cell apparatus. The wafer was placed in the donor compartment of the Franz cell on a wire mesh used as a membrane and the receptor compartment was filled with PBS (pH 6.8±0.1) solution until it just touched the wire gauze and the water jacket was filled with distilled water. The apparatus was placed on a heated magnetic stirrer and kept at 37°C. At set intervals of 15, 30, 45, 60, 90 and 120 min, 1ml aliquots were removed and placed in plastic tubes. To maintain a consistent volume, 1ml of buffer solution was added after withdrawal of each aliquot. Each sampled aliquot was then treated with 2ml Bradford's reagent and absorbance measured at 595nm. The calibration curve was used to calculate the concentration of BSA at each sample time point, and used to plot a graph of percentage drug release against time.
Results and Discussion
Pre-formulation studies
Ideal gels (1% w/w) were selected on the basis of ease of pouring and homogeneity with no lumps of undissolved polymer. Preliminary investigations involved the use of different concentrations of acetic acid (0.05%, 0.10%, 0.25%, 0.5% and 1%, and v/v) to prepare Ch gels. This was due to the fact that unlike SA, Ch which is the deacetylated derivative of chitin is insoluble in deionised water. However, it has free proton able amino groups present in the D-glucosamine unit in its structure which makes it soluble in aqueous acidic conditions and also important for some of its known properties [31, 32]. However, acetic acid concentration of 1 % w/w posed a potential risk with regards to toxicity without removing residual traces. Therefore, the different lower concentrations were used to prepare Ch gels. Based on visual observations, it was concluded that all the gels appeared similar with complete dissolution of the Ch as shown in Figure 1 below. As a result, the lowest acetic acid concentration (0.05 % v/v) was selected for all subsequent gel preparation as that was deemed safest for buccal application as well as not requiring the need for any further purification step such as membrane dialysis.

Figure 1: Ch gels produced using different concentrations of acetic acid (a) 0.05, (b) 0.10, (c) 0.15, (d) 0.25, (e) 0.50 and (f) 1.0 % v/v showing clear transparent solutions at each concentration of acetic acid.
The composite gel comprising SA and Ch in the ratio of ratio 4:1 was chosen as the ideal combination as it was free flowing and therefore easy to pour and remained clear and transparent. The other combined gels were too viscous and easily set to a solid mass, which made stirring difficult and also difficult to pour for freeze-drying. This increase in viscosity with increasing Ch content could be related to electrostatic interactions between the manuronate / guluronate side chains (negatively charged) of SA and the protonated amine group of Ch which is expected to increase with increasing chitosan content resulting in a stronger polymeric gel structure.
Differential scanning calorimetry (DSC)
DSC provided vital information such as the glass transition and eutectic temperature for the polymeric gels and is summarized in Table 2 below. This was important for determining the thermal events occurring during thermal treatment (freezing and subsequent heating) to determine the optimum conditions for freeze-drying (glass transition and eutectic melts), to avoid possible product collapse during the primary drying stage.
![]()
7
Glass transition peak
(°C)
Melt peak
(°C)
SA powder
41.54
-
Ch powder
-
-
SA gel
-
4.18
Ch gel
-
2.76
SA: Ch 4:1 gel
0.64
Table 2: DSC profiles of major peaks during thermal treatment simulating that occurring during freeze-drying.
Freeze drying
Freeze-drying relies on two principles, temperature and pressure. During the freeze-drying process the polymeric gel was first frozen to form a solute-ice crystal mixture referred to as freeze-concentrate. In the second phase, heat is applied at a low pressure which allows the ice crystals in the freeze-concentrate to be sublimated without going through the liquid phase. This ensured the water in the polymer was completely removed leaving behind a porous matrix in the form of a wafer. Generally, the blank and BSA loaded wafers were creamy white in appearance and spongy in nature due to the air pockets locked within their porous matrix structure. However, addition of the BSA made the wafers tougher possibly due to the interaction with the polymer. Figure 2 shows digital images of the lyophilised wafers prepared from the various gels described above, after the freeze-drying process. From visual observation, the wafer obtained by pouring 5g of gel was not deemed feasible to be used as a buccal delivery system because of relatively large thickness which will make it difficult to retain in the cheek region without biting into it. These will most likely cause discomfort and reduce patient compliance. On the other hand, the wafers obtained from pouring 1 and 2 g of gel were too thin to be effective as it will most likely disintegrate rapidly with saliva washing. As a result, the wafer obtained from pouring 3g of gel was the formulation of choice for drug loading.
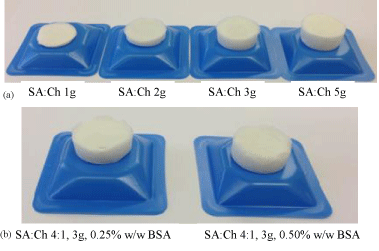
Figure 2: (a) Freeze-dried wafers obtained by pouring different weights of composite (SA:Ch 4:1) gels showing differences in thickness (b) BSA loaded wafers obtained from pouring 3g of SA:Ch 4:1 gels.
Physical characterisation
Hardness Test
Table 3 shows the results from compressing the wafers representing the peak resistance force to deformation to a given depth of compression. The results for the blank wafers showed a gradual increase in hardness with increasing thickness (weight of gel poured) though the differences were not marked. The increase could relate to relatively higher polymer density with increasing weight of gel poured as the higher weights contained more polymers per unit volume of wafer [33] and could well be related to changes in porosity, though this requires investigation. The results for the drug-loaded wafers show that formulations containing 0.50 % w/w BSA were stronger than the blank wafers and those containing 0.25% w/w BSA. Generally, a hardness value between 3 - 6 N is ideal [33], however, this is dependent on other factors such as the type of polymer used and the intended application. For example, wafers meant for rapid release will require a relatively low hardness value to allow rapid hydration, disintegration and subsequent drug release. For a fixed dose of macromolecule, the hardness could be adjusted by changing the total concentration of polymer or by incorporation of an appropriate plasticizer. The former will increase the hardness value whilst the latter generally decreases the rigidity of the polymer matrix with a resultant decrease in hardness. The increase in hardness for BSA loaded wafers could also be due to the interaction between the protein and the two polymers used. The possible interactions may vary between the polymers (SA or Ch) and the BSA and also complicated by the interaction between the two polymers themselves. For example, the amine functionality of Ch and hydroxyl groups of both SA and Ch can couple with the BSA [34]. Hydrophobic interactions between the non polar regions of the polymers and the BSA might also be possible. Strong charge to charge interactions have also been demonstrated between proteins and Ch and known to affect some of its physical characteristics [35]. However, these require further investigation to determine the specific interactions involved.
![]()
BLANK
Wafers prepared from pouring different weights of gel
Force (N)
1
2
3
Average
1g
2.30
2.42
2.40
2.37
2g
2.62
2.72
2.62
2.65
3g
2.90
2.724
2.83
2.82
5g
2.90
2.94
2.98
2.94
Wafers containing different amounts of BSA (% w/w)
DRUG LOADED
1
2
3
Average
0.25
4.30
4.53
4.34
4.39
0.50
6.02
5.47
5.69
5.73
Table 3: Resistance to deformation (hardness) of blank and drug loaded wafers prepared from SA:Ch 4:1 gels showing the effect of wafer thickness and BSA concentration. The drug loaded wafers were prepared by pouring 3 g of gel.
Swelling Capacity
Figure 3a shows the swelling profiles for blank wafers prepared by pouring 1, 2, 3 and 5 g of the combined gel (SA: Ch 4:1.). From the results, it can be observed that all of the blank wafers reached full saturation in 5 min, and the downward profile after 5 min represents the gradual erosion of the wafers. The wafers obtained from pouring 1 g and 2g of gel reduced significantly in weight after 15minutes. This suggests that at 15 minutes, the wafer began to completely disintegrate corresponding to the loss of structural integrity. However, the wafer obtained from pouring 5g of gel retained its structural integrity owing to the slower rate of hydration and swelling, due to the presence of higher amount of total polymer within the same volume of matrix.
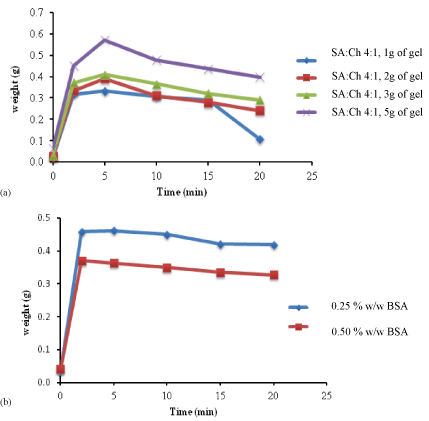
Figure 3: (a) profiles for blank wafers prepared by pouring 1, 2, 3 and 5 g of the combined gel (SA: Ch 4:1) and (b) swelling profiles of drug loaded wafers prepared from pouring 3g of combined gel (SA: Ch 4:1).
The drug loaded wafers reached saturation in 2 min, but unlike the blank wafers, the swelling capacity remained fairly constant implying that the drug loaded wafers maintained their structural integrity over a longer time period, and lost minimal weight in comparison to the blank wafers which all showed signs of disintegration. This may be due to the bonding between the BSA and the polymer as suggested above. Further, the wafers containing higher amounts of BSA showed slightly lower swelling but showed a more constant weight loss after 2 min compared to the 0.25% BSA loaded wafer which showed a much more rapid weight reduction after 10 min.
Scanning electron microscopy (SEM)
Figure 4 shows the SEM images for blank and drug loaded wafers all showing a porous polymeric network within the wafer matrix. The blank wafers obtained from pouring 1g of gel appear to show smaller pores that were more uniformly distributed compared to those obtained from pouring 2g and 3g of gel. This is quite interesting, given that there was less polymer present in the former (1 g of gel). The drug loaded wafers appeared flakier and brittle in appearance compared to the blank wafers and might be related to the presence of BSA dispersed on the polymer strands within the wafer matrix. This could also explain the reason for the increased resistance to deformation ('hardness') of the BSA loaded wafers when compared with the blank wafers.

Figure 4: SEM images of optimised blank and drug loaded wafers obtained from SA: Ch 4:1 combined gels showing their morphological microstructure.
Mucoadhesion
Figure 5 shows that wafers prepared from pouring 2 and 3 g of gel had similar peak adhesive force (PAF) values. However, addition of BSA increased the PAF implying that adhesion was proportional to the amount of BSA present in the wafers up to 0.50 % w/w. This is vital as the functional performance of the wafer relate to how well and how long the wafer would adhere to the lining of the cheek and the overall release of the drug. Given the improved mucoadhesion behavior with BSA loading, it is expected that this will facilitate the absorption of macromolecules such as BSA via the buccal mucosa which will allow improved bioavailability in the absence of a permeation enhancer as was the case in this study. This will however, require further investigations with an ex vivo or in vivo permeation study.
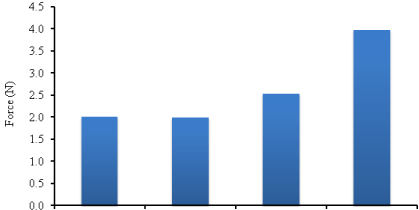
Figure 5: Mucoadhesion profiles of optimised blank and drug loaded combined wafers showing PAF values.
X-ray diffraction (XRD)
The diffractogram (Figure 6) shows a variety of peaks, relating to the nature of the compound. From Figure 6, it is evident that BSA and SA powder are amorphous in nature as shown by the broad peaks in the diffractogram. Ch shows a characteristic intense peak around 23 two theta, which was also present in the blank and drug loaded wafers, suggesting some level of crystallinity. However, this could be related to other salts naturally present in Ch. Overall, it appears that the drug maintained its amorphous nature within the final wafers and implies it did not change its form during gel formulation in combination with SA as well as during freeze-drying. Stable amorphous forms are advantageous as they exhibit better solubility and therefore higher rates of dissolution which is expected to enhance drug release and subsequent absorption and bioavailability. However, though the amorphous drug was maintained, XRD cannot confirm protein conformational stability which will need to be confirmed using circular dichroism and mass spectrometry.
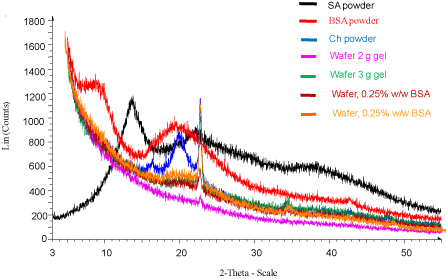
Figure 6: XRD diffractograms of pure starting materials, optimised blank wafers and drug loaded wafers.
In vitro drug dissolution study
Figure 7a shows the calibration curve used to determine the amounts of BSA released from the wafers at each time point. The dissolution graphs (Figure 7b) show a sustained release of BSA over a two hour period. The rate of release was faster from the wafer containing 0.25% w/w BSA and this formulation also showed a higher total % cumulative drug release after two hours compared to the formulation containing the higher amount of BSA with 66 % and 31% cumulative release respectively. These observations are interesting as the drug dissolution profiles directly mirror that observed in the hydration and swelling profiles of the two formulations. For swellable release systems such as SA and Ch, drug release is dependent on the rate of matrix hydration and subsequent diffusion of dissolved drug from the swollen polymer in the initial stages of drug release. Eventually, drug release is controlled by a combination of polymer swelling, drug diffusion and polymer erosion [36] which is possible in the current formulations.
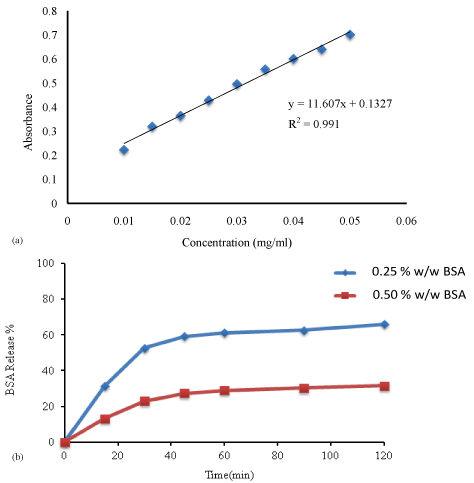
Figure 7: (a) Calibration curve of BSA (b) dissolution profiles of drug loaded wafers showing release of BSA from wafers containing different amounts of drug.
Though the proposed composite buccal drug delivery formulation has potential for administration of macromolecules, there are some challenges in terms of eventual clinical applications. The current study used BSA as a model drug, however, use of therapeutic peptides such as insulin and larger proteins such as growth hormone are required in relatively small quantities which can be difficult to formulate and scale up. Furthermore, buccal delivery of macromolecules will not be rapid compared to parenteral injections and not easily administered for comatose patients and very young children.
Conclusion
A lyophilized composite SA and Ch wafer loaded with BSA has been developed and optimized. The optimum wafer comprised SA and Ch in a 4:1 ratio respectively. The optimized formulations showed excellent functional mucosal characteristics including hardness, mucoadhesion, and swelling and in vitro drug release. The release of BSA which remained amorphous in the final wafer followed a sustained type release attributed to the rate of hydration and swelling. The optimized composite formulation offers a potentially effective drug delivery system for buccal administration of large macromolecules.
References
- Ayensu I, Mitchell JC, Boateng JS . Effect of membrane dialysis on characteristics of lyophilised chitosan wafers for potential buccal delivery of proteins. Int J Biol Macromol. 2012; 50: 905-909.
- Kianfar F, Antonijevic M, Chowdhry B, Boateng JS . Lyophilized wafers comprising carrageenan and pluronic acid for buccal drug delivery using model soluble and insoluble drugs. Colloids Surf B Biointerfaces. 2013; 103: 99-106.
- Kianfar F, Ayensu I, Boateng JS . Development and physico-mechanical characterization of carrageenan and poloxamer-based lyophilized matrix as a potential buccal drug delivery system. Drug Dev Ind Pharm. 2014; 40: 361-369.
- Giovino C, Ayensu I, Tetteh J, Boateng JS . An integrated buccal delivery system combining chitosan films impregnated with peptide loaded PEG-b-PLA nanoparticles. Colloids Surf B Biointerfaces. 2013; 112: 9-15.
- McInnes FJ, Thapa P, Baillie AJ, Welling PG, Watson DG, Gibson I, et al. In vivo evaluation of nicotine lyophilised nasal insert in sheep. Int J Pharm. 2005; 304: 72-82.
- Pawar HV, Tetteh J, Boateng JS . Preparation, optimisation and characterisation of novel wound healing film dressings loaded with streptomycin and diclofenac. Colloids Surf B Biointerfaces. 2013; 102: 102-110.
- Pawar HV, Boateng JS, Ayensu I, Tetteh J . Multifunctional medicated lyophilised wafer dressing for effective chronic wound healing. J Pharm Sci. 2014; 103: 1720-1733.
- das Neves J, Bahia MF . Gels as vaginal drug delivery systems. Int J Pharm. 2006; 318: 1-14.
- Boddupalli BM, Mohammed ZN, Nath RA, Banji D . Mucoadhesive drug delivery system: An overview. J Adv Pharm Technol Res. 2010; 1: 381-387.
- Smart JD . The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005; 57: 1556-1568.
- Boateng JS, Ayensu I . Preparation and characterization of laminated thiolated chitosan-based freeze-dried wafers for potential buccal delivery of macromolecules. Drug Dev Ind Pharm. 2014; 40: 611-618.
- Patel VF, Liu F, Brown MB . Advances in oral transmucosal drug delivery. J Control Release. 2011; 153: 106-116.
- Shojaei AH . Buccal mucosa as a route for systemic drug delivery: a review. J Pharm Pharm Sci. 1998; 1: 15-30.
- Zhang H, Zhang J, Streisand JB . Oral mucosal drug delivery: clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 2002; 41: 661-680.
- Ayensu I, Mitchell JC, Boateng JS . Development and physico-mechanical characterisation of lyophilised chitosan wafers as potential protein drug delivery systems via the buccal mucosa. Colloids Surf B Biointerfaces. 2012; 91: 258-265.
- Harris D, Robinson JR . Drug delivery via the mucous membranes of the oral cavity. J Pharm Sci. 1992; 81: 1-10.
- Bernkop-Schn�rch A, Kast CE . Chemically modified chitosans as enzyme inhibitors. Adv Drug Deliv Rev. 2001; 52: 127-137.
- Giovino C, Ayensu I, Tetteh J, Boateng JS . Development and characterisation of chitosan films impregnated with insulin loaded PEG-b-PLA nanoparticles (NPs): a potential approach for buccal delivery of macromolecules. Int J Pharm. 2012; 428: 143-151.
- Boateng JS, Mitchell JC, Pawar H, Ayensu I1 . Functional characterisation and permeation studies of lyophilised thiolated chitosan xerogels for buccal delivery of insulin. Protein Pept Lett. 2014; 21: 1163-1175.
- Ayensu I, Boateng JS. Development and evaluation of lyophilized thiolated-chitosan wafers for buccal delivery of protein. J Science Technol. 2012; 32: 46-55.
- Funami T, Fang Y, Noda S, Ishihara S, Nakauma M, Draget KI, et al. Rheological properties of sodium alginate in an aqueous system during gelation in relation to supermolecular structures and calcium binding. Food Hydrocoll. 2009; 23(7): 1746-1755.
- Fan L, Jiang L, Xu Y, Zhou Y, Shen Y, Xie W, et al. 2001. Synthesis and anticoagulant activity of sodium alginate sulfates. Carb Polym. 2001; 83: 1797-1803.
- Wu ZM, Zhang XG, Zheng C, Li CX, Zhang SM, Dong RN, et al. Disulfide-crosslinked chitosan hydrogel for cell viability and controlled protein release. Eur J Pharm Sci. 2009; 37: 198-206.
- Muzzarelli RAA, Boudrant J, Meyer D, Manno N, DeMarchis M, Paoletti MG. Current views on fungal chitin/chitosan, human chitinases, food preservation, glucans, pectins and inulin: A tribute to Henri Braconnot, precursor of the carbohydrate polymers science, on the chitin bicentennial. Carb Polym. 2012; 87: 995-1012.
- Xu Y, Zhan C, Fan L, Wang L, Zheng H . Preparation of dual crosslinked alginate-chitosan blend gel beads and in vitro controlled release in oral site-specific drug delivery system. Int J Pharm. 2007; 336: 329-337.
- Janes KA, Calvo P, Alonso MJ . Polysaccharide colloidal particles as delivery systems for macromolecules. Adv Drug Deliv Rev. 2001; 47: 83-97.
- George M, Abraham TE . Polyionic hydrocolloids for the intestinal delivery of protein drugs: alginate and chitosan--a review. J Control Release. 2006; 114: 1-14.
- Lanjhiyana SK, Bajpayee P, Kesavan K, Lanjhiyana S, Muthu MS. Chitosan-sodium alginate blended polyelectrolyte complexes as potential multiparticulate carrier system: colon-targeted delivery and gamma scintigraphic imaging. Expert Opin Drug Deliv. 2013; 10: 5-15.
- Li P, Dai YN, Zhang JP, Wang AQ, Wei Q . Chitosan-alginate nanoparticles as a novel drug delivery system for nifedipine. Int J Biomed Sci. 2008; 4: 221-228.
- Shaikh AA, Pawar YD, Kumbhar ST. Effect of Chitosan and Sodium Alginate on Mucoadhesion and Drug Release of Itraconazole Tablets. Int J Res Pharm Biomed Sci. 2012; 3: 293 - 297.
- Shaikh AA, Pawar YD, Kumbhar ST. Effect of Chitosan and Sodium Alginate on Mucoadhesion and Drug Release of Itraconazole Tablets. Int J Res Pharm Biomed Sci. 2012; 3: 293 - 297.
- Romanazzi G, Gabler FM, Margosan D, Mackey BE, Smilanick JL . Effect of chitosan dissolved in different acids on its ability to control postharvest gray mold of table grape. Phytopathology. 2009; 99: 1028-1036.
- Boateng JS, Auffret AD, Matthews KH, Humphrey MJ, Stevens HN, Eccleston GM . Characterisation of freeze-dried wafers and solvent evaporated films as potential drug delivery systems to mucosal surfaces. Int J Pharm. 2010; 389: 24-31.
- Yang L, Hsiao WW, Chen P. Chitosan-cellulose composite membrane for affinity purification of biopolymers and immunoadsorption. Journal Membrane Science. 2002; 197: 185-197.
- Ma Z, Yeoh HH, Lim LY . Formulation pH modulates the interaction of insulin with chitosan nanoparticles. J Pharm Sci. 2002; 91: 1396-1404.
- Boateng JS, Matthews KH, Auffret AD, Humphrey MJ, Eccleston GM, Stevens HN . Comparison of the in vitro release characteristics of mucosal freeze-dried wafers and solvent-cast films containing an insoluble drug. Drug Dev Ind Pharm. 2012; 38: 47-54.