
Research Article
Austin Biomark Diagn. 2016; 3(1): 1024.
The Complete Spectrum of Beta (β) Thalassemia Mutations in Bangladeshi Population
Sultana GNN1*, Begum R1, Akhter H2, Shamim Z3, Rahim MA4 and Chubey G5
1Centre for Advanced Research in Sciences, University of Dhaka, Bangladesh
2Bangabandhu Sheikh Mujib Medical University (BSMMU), Bangladesh
3Popular Diagnostic Center, Dhanmondi, Dhaka, Bangladesh
4Thalassemia Foundation Hospital, Chamelibag, Bangladesh
5Estonia Biocenter, Tartu, Estonia
*Corresponding author: Sultana GNN, Centre for Advanced Research in Sciences, University of Dhaka, Bangladesh,
Received: March 18, 2016; Accepted: April 06, 2016; Published: April 12, 2016
Abstract
Background: In Bangladesh, 3% people are carriers of β-thalassemia and 4% people are E- β-thalassemia. However to date, very little data about HBB gene mutations in the population of Bangladesh are available. This study was done to determine the complete spectrum of β-thalassemia mutations in the population of Bangladesh. We analyzed the entire HBB gene including regulatory regions in 19 unrelated β-thalassemia major and 51 E- β-thalassemia patients and 33 healthy controls using DNA direct sequencing method.
Result: Nine β-thalassemia polymorphisms were identified in Bangladeshi thalassemia patients. The most common polymorphisms were IVSI -5 G > C (81.4%), CD 26/ HbE: G>A (72.85%), CD 2: T>C (57.1%) and IVSII-16: G>C (57.1%). The less common polymorphisms were -90 C>T (1.43%), CD1 T>A (2.86%), CD2 C>A (5.71%), CD30 G>C (1.43%), and IVS-II-81 C>T (1.43%). These polymorphisms were distributed as 6 genotyping patterns in β-thalassemia major and 8 in E- β-thalassemia patients. IVS1-5: G>C alone or combined with other β thalassemia polymorphisms are responsible for pathogenic condition in β-thalassemia major and along with CD26/E: G>A and other β thalassemia mutations in most E- β-thalassemia. CD2: T>C alone cannot produce pathogenic condition but when it combined with CD1: T>A and CD2: C>A it can generated thalassemia. This CD2: T>C and another common polymorphism IVSII-16: G>C along with IVSII-74: T>G were also identified in healthy controls. Altogether 17 haplotypes among the Bangladeshi populations was observed and two shared haplotypes among patients and controls. The haplotype diversity was substantially higher among patients and lower among controls.
Conclusion: The genotypic profile of β—thalassemia in Bangladesh patients shows great variability. IVS1-5: G>C most common polymorphism for β- thalassemia and a combination of IVS1-5: G>C and CD26/E: most common polymorphism for E β-thalassemia disease in Bangladeshi population.
Keywords: β-thalassemia; Mutations; Bangladesh; Heterozygous; Homozygous
Introduction
β-thalassemia is a genetic disorder that is prevalent in certain parts of the world including Bangladesh [1,2]. The incidence for this disease is high in tropical and subtropical areas including Southeast Asia [3]. In Bangladesh the carrier rate of β-thalassemia is 3.0% and Hb-E/β- thalassemia is 4.0% and affected birth per thousand of β-thalassemia and Hb-E/ β-thalassemia is 0.106 & 3.000 respectively [4]. About 10% of the world’s thalassemia major children are born in India [5]. Gene flow from Indian sub-continent to Bangladesh may be one of the reasons of prevalence of this disease in Bangladesh [5-6]. Other reasons of prevalence of thalassemia disease are intermarriage between different ethnic groups, lack of awareness for blood test before marriage [7]. Besides accurate data on carrier rates in Bangladesh is lacking, insufficient technologies for prenatal diagnosis also increases the prevalence of beta thalassemia in Bangladesh.
The function of hemoglobin in human and animal is very important which carries oxygen from lungs to other parts of the body. It is metalloprotein having quaternary structure which contains iron and performs the important function of transporting oxygen via RBCs in blood in mammals as well as other animals [8]. It also effect modulation and gas transport duties, although it differ from species to species and most probably is altogether different in vertebrates. As long as more oxygen is bound to hemoglobin more oxygen is reached to every part of the body. The external chemical factor which helps in regulation of oxyhemoglobin affinity induces, pH, 2, 3-diphospahoglycerate and carbon dioxide.
β-thalassemia can be divided into three main types depends on the clinical phenotypes: thalassemia major, thalassemia trait and thalassemia intermediate. β-thalassemia major is a severe form that requires transfusions from infancy for survival. This clinical phenotypic variability β-thalassemia occurs due to the mutation in three exons and two intervening sequences, 5´ UTR (untranslated region) and 3´ UTR of β-globin (HBB) gene [9-10]. Mutation in exons and intervening sequences that may produces non-functional beta globin protein or β0 allele and mutation in HBB promoter, 5´ UTR and 3´ UTR that may produces reduced quantity of beta globin protein or β+ allele [10,11]. Approximately 600 mutations have been identified in the β-globin (HBB) gene, of which more than 200 are associated with β-thalassemia phenotype [11,12]. The distribution and frequency of different mutations regarding thalassemia vary from population to population [13-15]. Haemoglobin E (HbE) allele, point mutation (G > A) in codon 26 of β - globin gene, can induce alternative splicing and thus result in decreased β -globin E chains [16]. Hb E/β-thalassemia results from co-inheritance of a β-thalassemia allele from one parent and Haemoglobin E from the other [17]. HbE/ β thalassemia causes a surprisingly variable anaemia, ranging from nearly asymptomatic states to severe anaemia and transfusion-dependency [18].
However, to date, the genetic basis of β- thalassemia in Bangladeshi patients is still poorly understood. Here, we attempt to identify the complete spectrum of β-thalassemia mutations in Bangladeshi population. To acquire the data we sequenced entire 1.6 kb HBB gene, 163 bp upstream and 153bp downstream regulatory region in 70 patients with β-thalassemia or Hb-E/ β-thalassemia phenotypes and 33 healthy controls. During the analysis, nine point mutations were identified, of them IVSI -5G > C in β-thalassemia major and CD26 (G-A) + IVSI -5G > C in Hb-E/β-thalassemia were frequent. Therefore our findings could provide genetic insight of beta thalassemia disease occurrence in Bangladeshi population and are useful in genetic counseling, diagnostic application and treatment.
Materials and Methods
Subjects
As Bengali people are less diverse, mutations of β-thalassemia are not regional specific [19]. Therefore, a total of 70 patients with β-thalassemia were included in this study came from different states of Bangladesh who visited Bangladesh Thalassemia Hospital for treatment after diagnosis. Samples were randomly selected from thousand and fourty five cases of our previous study [20]. Out of them 19 patients were β-thalassemia major and 51 were HbE-β- thalassemia. 33 healthy control samples were also chosen to evaluate the mutations profile of β-thalassemia in Bangladesh. Blood samples were collected from the patients under an agreement with the Head of the Bangladesh Thalassemia Hospital with Centre for Advanced Research in Sciences (CARS) and Department of Biochemistry and Molecular Biology, University of Dhaka. The protocol for this study was approved by ethics committee of Bangladesh Medical Research Council (BMRC). Informed consent was obtained from each individual or parents of individuals younger than 18 years old. Approximate 3-5 ml of blood sample was collected in EDTA coated vacutainer from both patients and controls and finally stored at -20o C until analysis.
Hematological analysis and Clinical data collection
Hematological analyzer (Sysmex XE-2100) was used to determine the complete blood counts and red blood cells. The data of Mean Corpuscular Volume (MCV) and Mean Corpuscular Haemoglobin (MCH) were evaluated. The Hemoglobin Electrophoresis was carried out by using Sebia’s Capillary Electrophoresis System (CAPILLARYS™ 2) and the levels of HbA, A2 and F were analyzed according to Sultana et al. [20]. Clinical data e.g. the information about blood transfusions was obtained by retrospective clinical data.
Beta (β)-Thalassemia mutations analysis
Genomic DNA was extracted from whole blood by a standard procedure of phenol/ chloroform/ isoamyl alcohol extraction [21]. The quality and quantity of extracted DNA was measured by Nano Drop spectrophotometer 2000 and visualized by 0.8% agarose gel electrophoresis in 1×TAE buffer.
A total of 1922 bp genomic sequence containing entire HBB gene, 163 bp upstream and 153bp downstream regulatory region was amplified and sequenced in 70 patients and 33 controls. This region was amplified using three pairs of primers and sequenced by these six end primers that were designed using Primer 3 software (https://frodo. wi.mit.edu/primer3/). The sequence of the primers will be available on request from the authors. Amplifications were performed in a 20 ml volume containing one unit of AmpliTaq Gold (Promega, USA), 1× Polymerase Chain Reaction (PCR) buffer, 1.87 mM MgCl2, 200 μM deoxynucleotides triphosphates, 5 pmol each of forward and reverse primer and 40-50ng of genomic DNA. The PCR program consisted an initial denaturation at 95° C for 10 min, followed by 35 cycles of 1 min at 950 C, 1 min at 600 C, 1 min at 720 C and with a final elongation at 720 C for 10 min. PCR products were visually verified on 1% agarose gels and directly sequenced using a Big Dye Terminator cycle sequencing kit V3.1 (Applied Biosystems, Foster City, CA) and by ABI PRISM® 3130 Genetic analyzer (Center for Advanced Research in Sciences or CARS, Dhaka University, Bangladesh). Each sequence was reconfirmed by second run of sequencing.
The chromatograms generated from the genetic analyzer along with the base sequences were analysed by Bio-edit Sequence Alignment editor (V 7.0) [22] and Mac-based software (Auto Assembler V 3.0). Patient’s sequences were compared with National Centre for Biotechnology Information (NCBI) RefSeq entry of HBB (NG_000007.3) using NCBI BLAST (bl2seq) tools. HbVar database was used for the identification of the presence of reported mutations in other populations [23].
Statistical analysis
Data were expressed as mean (±SD) and number (percentage) as appropriate. The mean (±SD) of each haematological and clinical parameter were calculated using Microsoft Excel 2010. The statistical analysis was of these data was calculated by using GraphPad statistical software (https://www.graphpad.com/quickcalcs/index.cfm ).
Statistical significance was determined by one way Analysis Of Variance (ANOVA). P value < 0.05 was considered to be statistically significant. We have used phased the data by using DNASP software [24]. The haplotypes were used to construct the Network (Fluxus Engineering). Arlequin 3.5 [25] was used to measure diversity indices.
Results
Basic clinical characteristics of β thalassemia patients of Bangladesh
The basic clinical characteristics of β thalassemia patients of Bangladesh revealed that there were no significant differences between patients with β thalassemia major and E-β thalassemia regarding WBC, RBC, Hb level, MCV, MCH and Body Mass Index (BMI) (Table 1). There were also no significance difference was found in time of blood transfusion taken from and frequency of blood transfusion (Table 1).
![]()
Haematological and clinical data analysis
Parameter
Beta thalassemia major
E-beta thalassemia
p-value
WBC(10^3/µL)
14.86 ±4.48
13.16±6.73
0.314
RBC(10^6/µL)
3.68±0.88
3.44 ±0.94
0.336
Hb level (g/dL)
7.4 ±1.67
6.9 ±1.66
0.267
MCV (fl)
71.88 ±10.63
69 ±9.8
0.514
MCH (pg)
21.36 ±2.51
19.94± 3.34
0.4734
Transfusion taken from
3 months to 4 year, 2 year is most frequent
8 months to 9 year, one patient also from one months
-
Frequency of transfusion
68% taken 1 time per month and other are taken randomly
64.24% taken 1 time per month and other are taken randomly
-
BMI
17.74±4.37
17.28±3.48
0.9475
Table 1: Haematological and clinical data of β-thalassemia major and E-beta thalassemia patients.
Beta (β)-thalassemia Mutations in Bangladeshi Population
In this study, nine mutations (-90 C>T, Codon 1 T>A, Codon 2 C>A, Codon 2 T>C, HbE /Codon 26 G>A, Codon 30 G>C, IVS-I-5 G > C, IVS-2-16 G>C and IVS-2-81 C>T) of HBB gene were found in the 70 Bangladeshi β thalassemia patients (Table 2). Among them, the most common mutation was IVS-I-5 G>C (81.4%). This result corresponds to previous studies that also found IVS-I-5 G > C to be the most common mutation in the Bangladeshi β- thalassemia patients.
![]()
Mutation
N0
Common name
Unique Identification
HGVS code
Frequency (%)
location within gene
Patients
Healthy controls
1
-90(C-T)
HBB:c.-140C>T
rs34999973
Promoter
1.43
-
2
CD1(T-A)
HBB:c.5T>A
rs33949930
Exon 1
2.86
-
3
CD2(C-A)
HBB:c.7C>A
rs35906307
Exon 1
5.71
-
4
CD2(T-C)
HBB:c.9T>C
rs713040
Exon 1
57.1
42.42
5
CD26(G-A) /HbE
HBB:c.79G>A
rs33950507
Exon 1
72.85
-
6
CD30(G-C)
HBB:c.92G>C
rs33960103
Exon 1
1.43
7
IVS-I-5(G-C)
HBB:c.92+5G>C
rs33915217
Intravenous sequence 1
81.4
-
8
IVS-II-16(G-C)
HBB:c.315+16G>C
rs10768683
Intravenous sequence 2
57.1
60.6
9
IVS-II-74(T-G)
HBB:c.315+74T>G
rs7480526
Intravenous sequence 2
-
33.33
10
IVS-II-81(C-T)
HBB:c.315+81C>T
rs7946748
Intravenous sequence 2
1.43
-
Table 2: Identified β thalassemia mutations in thalassemia patients and healthy controls of Bangladesh.
The second most common mutation was HbE /Codon 26 G>A (72.85%) and the third most common mutations were Codon 2 T>C (57.1%) and IVS-2-16 G>C (57.1%). Other five mutations (-90 C>T, Codon 1 T>A, Codon 2 C>A, Hb Monroe (Codon 30 G>C), and IVS- 2-81 C>T were less frequent (1.43% to 5.71%) in Bangladeshi patients (Table 2). Three HBB variants Codon 2 T>C (42.42%), IVS-2-16 G>C (60.60%) and IVS-2-74 T>G (33.33%) were identified in Bangladeshi healthy controls (Table 2). Among these, IVS2-16 G> C and Codon 2 T>C also identified in patients but IVS-2-74 T>G only identified in Bangladeshi healthy controls. The variants IVS-2-16 G>C and Codon 2 T>C are also common in Bangladeshi β-thalassemia patients (Table 2).
Genotypic pattern of Beta thalassemia mutations in Bangladeshi patients
Nine mutations were distributed in 14 genotypic patterns among Bangladeshi β thalassemia patients (Table 3). In thalassemia major patients, IVS1-5: G>C mutation alone can be formed pathogenic condition in homozygous genotype or it can be formed disease combined with other mutation in both homozygous and heterozygous genotype. In E-β thalassemia patients, 5 out of 8 mutational genotypic pattern also contained IVS1-5: G>C in heterozygous genotype along with CD26/E: G>A mutation and other beta thalassemia mutations. Another common mutation CD2: T>C in both homozygous and heterozygous genotype was found in 3 out of 5 genotypic pattern. This mutation was found most of healthy controls either in heterozygous or homozygous form. This SNP has no pathogenic effect when it form genotypic pattern with IVS2-16: G>C (Homo), IVS2-74: T>G (Homo), but when it combined with CD1: T>A (Hetero), CD2: C>A (Hetero), CD2: T>C (Homo), it have pathogenic effect. Another most common mutation IVS-II-16:G>C was identified in only one β thalassemia major genotypic pattern in heterozygous form but in E- β-thalassemia it was found in 4 out of 8 in both homozygous and heterozygous form. Most interestingly this mutation was also found most of healthy controls either in heterozygous or homozygous form. Therefore it can be concluded that CD2: T>C and IVS-II-16: G>C might be only a polymorphism which has no pathogenic effect. Therefore it can be concluded that IVS1-5: G>C alone or combined with other β thalassemia mutation formed β-thalassemia major and combined with CD26/E: G>A formed E-β-thalassemia.
![]()
Thalassemia type
Genotypic pattern of b thalassemia mutations
Thalassemia major
IVS1-5: G>C (Homo)
IVS1-5: G>C (Homo), CD2:T>C (Homo)
IVS1-5: G>C (Homo), CD2:T-C (Hetero)
IVS1-5: G>C (Hetero), -90 C>T(Hetero), IVS2-81: C>T (Hetero)
CD1:T>A (Hetero), CD2:C>A (Hetero), CD2:T>C (Homo)
IVS1-5: G>C (Hetero), CD2:C>A (Hetero), CD2:T>C (Hetero), CD30:G>C (Hetero), IVS-II-16:G>C (Hetero)
E-beta thalassemia
CD26/E:G-A (Hetero ), IVS1-5: G>C (Hetero), CD1;T>A (Hetero),
CD2:C-A (Hetero), CD2:T>C (Hetero), IVS2-16: G>C (Hetero)
CD26/E:G>A (Hetero), IVS1-5: G>C (Hetero), CD2:T>C (Hetero),
IVS2-16: G>C (Hetero)
CD26/E:G>A (Hetero), IVS1-5: G>C (Hetero), CD2:T>C (Hetero), IVS2-16: G>C (Homo)
CD26/E:G>A (Hetero), IVS1-5: G>C (Hetero), CD2:T>C (Hetero)
CD26/E:G>A (Hetero), IVS1-5: G>C (Hetero),
CD26/E:G-A (Homo), CD2:T>C (Homo), IVS2-16: G>C (Homo)
CD26/E: G>A (Hetero), CD2: C>A (Hetero), CD2: T>C (Homo)
CD26/E: G>A (Hetero), CD2:T>C (Homo)
Table 3: Genotypic pattern of β-thalassemia mutations in Bangladeshi patients.
All together 17 haplotypes among the Bangladeshi populations was observed. The Network reconstruction showed two shared haplotypes among patients and controls, whereas numbers of haplotypes were substantially higher among cases (Figure 1). In this study, it was also found that the haplotype diversity was higher among patients and lower among controls (Figure 2). Therefore it can be concluded that the genotypic profile of βthalassemia in Bangladeshi beta thalassemia patients shows great variability. Tajima and Fu’s test also applied to detect the signal of selection. The result didn’t show any sign of selection.
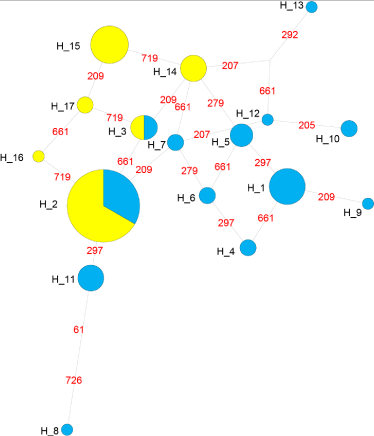
Figure 1: The median-joining network of disease and control haplotyes
obtained from the phased sequence data in present study. Circles sizes
are proportional to the number of chromosomes in a particular haplotype.
Network was drawn by the Network software (ver 4, https://www.fluxusengineering.
com/).
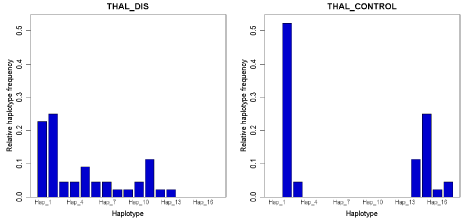
Figure 2: The comparison of diversity as well as frequency of hapltypes present in both case and control individuals. The haplotypes were identified from the
phased sequence data. The frequency of each haplotype was counted by the software Arlequin 3.5 (https://cmpg.unibe.ch/software/arlequin35/) and figure was
drawn by the script of R (https://cmpg.unibe.ch/software/arlequin35/) implemented in Arlequin.
Discussion
As a result of geographical position and global population movement, β-thalassemia becomes a common haemoglobin genetic disorder in Bangladesh. There are several reports on the spectrum of mutations in the Indian subcontinent and Middle East [26-28]. However, a very few data was found on the β- thalassemia mutations in Bangladeshi patients. One study has been carried out with limited number of samples from a segment of HBB gene [29]. This is the first report on detail of HBB gene mutations in Bangladeshi population on the basis of complete HBB gene with 163bp upstream and 153bp downstream regulatory regions. The present study also has analyzed a larger number of β-thalassemia genes from suspected β- thalassemia patients and healthy controls. Here we identified nine mutations in patients; of which only four mutations corresponded to previous report [29]. Therefore, the present study demonstrates that, β-thalassemia is heterogeneous in Bangladesh. Three mutations i.e. Codon 1 T>A, Codon 2 C>A and IVS-2-81 C>T were identified first time in Bangladeshi patients; which were nor found in any other geographically related countries [9,27,28].
IVS-I-5 G> C was found to be the most common identified mutation in Bangladeshi patients, occurring at percentages of 81.4. This mutation was also common (9 out of 16 and 13 out of 17) in previous reports [29-30]. Even in Indian population this mutation is most common with a frequency of 63.17%, in the UAE (55%) and in Oman (62%) [27,31]. Mutation in this position reduces splicing efficiency, resulting in aberrant β-globin chain formation [32]. HbE (Codon 26 G>A), the second most common mutation in this study with a Percent of 72.85 and most of the Hb-E/β-thalassemia patients had IVS-I-5 G> C mutation along with HbE (Codon 26 G>A). Ibn Ayub et al., [29] also observed 62.5% patients had this Codon 26 G>A mutation. HbE (Codon 26 G>A) produces variant haemoglobin along with a cryptic splice site, resulting in abnormal messenger RNA (mRNA) processing [16]. Hemoglobin E when co-inherited with a β-thalassemia mutation causes serious disease [33]. Codon 2 T>C and IVS-2-16 G>C were the third most common mutation in this study with a frequency of 57.1%. Codon 2 T>C changes CAT to CAC but both coding for Histidine. This mutation also documented in previous report [29]. IVS-2-16 G>C is in the alternative splicing region of mRNA, which might lead to differential expression. This polymorphism is quite common in Indian populations. All over India fifty-five populations that were studied have revealed this C allele frequency ranging from 0.21 in Indo-Europeans to 0.66 in Dravidian linguistic groups [9]. Codon 2 T>C and IVS-2-16 G>C mutations had no apparent effect in Bangladeshi thalassemia population because these were also found to be present in healthy controls with a Percent of 42.42 and 60.60 respectively.
The less common mutations have included -90 C>T (1.43%), Codon 1 T>A (2.86%), Codon 2 C>A (5.71%), Hb Monroe (Codon 30 G>C) (1.43%), IVS-2-74 T>G and IVS-2-81 C>T (1.43%). -90 C>T is a rare mutation, located in the proximal CACCC box of promoter region. This mutation is first described in Portuguese population [34]. This mutation has also been reported in China and India but not in Bangladesh [27]. The other rare mutation Hb Monroe (Codon 30 G>C) mutation found in only one patient that not only leads to undetectable mutant peptide and transcript but also interferes with the expression of wild allele [35]. This mutation has been previously reported in Bangladesh by Ibn Ayub et al. [29]. Codon 1 T>A (HBB: c.5T>A) was found in two patients in this study which changes the amino acid valine (GTG) to glutamic acid (GAG). The introduction of this glutamic acid residue prevents the removal of the initiator methionine, thus extending the N-terminus by one residue and it was first reported in three members of a Qatari family [36]. Codon 2 C>A (HBB: c.7C>A) was found in four patients in this study which changes the amino acid Histidine (CAT) to Asparagine (AAT). C>T substitution in this position of Codon 2, changes the amino acid Histidine (CAT) to Tyrosine (TAT) was previously reported [37]. 2nd base of this Codon HBB: c.8A is replace by C,G, T and change the amino acid Histidine to Proline (CCT), Arginine (CGT) and Leucine (CTT) respectively that altered the 2,3-diphosphoglycerate binding site. Therefore, the previous reports verify the significance of these mutations (Codon 1 T>A and Codon 2 C>A) in the pathogenesis in Bangladesh. IVS-2-74 T>G was found only in healthy controls. This mutation was previously reported in in Karnataka, India [9] and also reported as a neutral sequence changes in the human beta globin gene [38]. Another mutation IVS-2-81 C>T observed in one patient along with IVS-1-5 G>C and -90 C>T /HBB: c.-140C>T which was not previously found in geographically related populations [27].
The haplotype based analysis suggested a clear difference of haplotypes of case and controls (Figure 1 and 2). Only, two of the haplotypes were shared, whereas cases had higher number of haplotypes. The controls showed a drastic reduction of diversity (Figure 2). However, surprisingly the test of selection was not significant suggesting that either the phenomenon is very recent or we didn’t have enough power to detect in the present analysis [39].
Conclusion
Therefore, from the result it can be concluded that the mutation IVS-1-5 G>C is the most common for the occurrence of β-thalassemia among the Bangladeshi population as Indian Subcontinent and South East Asia. HbE (Codon 26 G>A) is another most common mutation in Bangladeshi patients. Some rare mutations included (-90 C>T, Codon 1 T>A, Codon 2 C>A, Hb Monroe / Codon 30 G>C and IVS- 2-81 C>T) are also responsible for the pathogenesis of β-thalassemia among the Bangladeshi population. Most interestingly, the identified mutations in this study and previous study [29] were located within HBB: c-92 to IVS-2-81 that means this location covered the 42 bp promoter, 5’UTR, exon 1, intravenous sequence 1, Exon 2 and 81 bp of intravenous sequence 2 of HBB gene. Thus this location might be used as biomarker for carrier-screening, for genetic counseling and for establishment of a comprehensive allele specific prenatal diagnosis kit for detection of β- thalassemia in Bangladesh.
Acknowledgements
We are thankful to all donors/patients for participating in this study. Special thanks to M. Faiyaz-Ul-Haque for giving us suggestion regarding the disease mutations. Authors are thankful to laboratory technicians M. Ashraful Islam and M. Moshiur Rahman. The authors are thankful to Third World Academy of Sciences (TWAS) for financial support to conduct this research.
References
- Weatherall D, Clegg JB. The Thalassemia Syndromes, Fourth ed. Blackwell Science Oxford. 2001.
- Uddin MM, Akteruzzaman S, Rahman T, Hasan AK, Shekhar HU. Pattern of β-thalassemia and Other Haemoglobinopathies: A Cross-Sectional Study in Bangladesh. ISRN Hematol. 2012; 2012: 659191.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998; 11: 1-51.
- Rahman MJ, Rahman MH. Prevention & control strategy of thalassemia in Bangladesh. The Orion Med. J. 2003; 16: 121-122.
- Sengupta M. Thalassemia among the tribal communities of India. The Internet Journal of Biological Anthropology. 2007; 1: 2.
- Gazi NN, Tamang R, Singh VK, Ferdous A, Pathak AK, Singh M, et al. Genetic structure of Tibeto-Burman populations of Bangladesh: evaluating the gene flow along the sides of Bay-of-Bengal. PLoS One. 2013; 8: e75064.
- Chen W, Zhang X, Shang X, Cai R, Li L, Zhou T, et al. The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity. BMC Med Genet. 2010; 11: 31.
- ones, Daniel (2003) [1917], Peter Roach, James Hartmann and Jane Setter, eds.,English Pronouncing Dictionary, Cambridge: Cambridge University Press, ISBN 3-12-539683-2
- Bilgen T, Clark OA, Ozturk Z, Akif Yesilipek M, Keser I. Two novel mutations in the 3' untranslated region of the beta-globin gene that are associated with the mild phenotype of beta thalassemia. Int J Lab Hematol. 2013; 35: 26-30.
- Kulkarni GD, Kulkarni SS, Kadakol GS, Kulkarni BB, Kyamangoudar PH, Lakkakula BV, et al. Molecular basis of β-thalassemia in Karnataka, India. Genet Test Mol Biomarkers. 2012; 16: 138-141.
- Vetter B, Neu-Yilik G, Kohne E, Arnold R, Sinha P, Gaedicke G, et al. Dominant beta-thalassaemia: a highly unstable haemoglobin is caused by a novel 6 bp deletion of the beta-globin gene. Br J Haematol. 2000; 108: 176-181.
- Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004; 124: 264-274.
- Huisman TH, Carver MF. The beta- and delta-thalassemia repository (Ninth Edition; Part I). Hemoglobin. 1998; 22: 169-195.
- Patrinos GP, Kollia P, Papadakis MN. Molecular diagnosis of inherited disorders: lessons from hemoglobinopathies. Hum Mutat. 2005; 26: 399-412.
- Agarwal S, Hattori Y, Agarwal SS. Rare beta-thalassemia mutations in Asian Indians. Am J Hematol. 2000; 65: 322-323.
- Tan JA, George E, Tan KL, Chow T, Tan PC, Hassan J, et al. Molecular defects in the beta-globin gene identified in different ethnic groups/populations during prenatal diagnosis for beta-thalassemia: a Malaysian experience. Clin Exp Med. 2004; 4: 142-147.
- Orkin SH, Kazazian HH Jr, Antonarakis SE, Ostrer H, Goff SC, Sexton JP. Abnormal RNA processing due to the exon mutation of beta E-globin gene. Nature. 1982; 300: 768-769.
- Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011; 134: 522-531.
- Fucharoen S, Winichagoon P. Prevention and control of thalassemia in Asia. Asian Biomed. 2007; 1: 1-6.
- Bandyopadhyay A, Bandyopadhyay S, Basak J, Mondal BC, Sarkar AA, Majumdar S, et al. Profile of beta-thalassemia in eastern India and its prenatal diagnosis. Prenat Diagn. 2004; 24: 992-996.
- Sultana GNN, Rahman A, Karim MA, Shahinuzzaman ADA, Begum R, Begum R. Breast cancer risk associated mitochondrial NADH-dehydrogenase subunit-3 (ND3) polymorphism. Journal of Medical Genetics and Genomics. 2011; 3: 131-135.
- Thangaraj K, Joshi MB, Reddy AG, Gupta NJ, Chakravarty B, Singh L. CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J Androl. 2002; 23: 815-818.
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999; 41: 95-98.
- Patrinos GP, Giardine B, Riemer C, Miller W, Chui DHK, Anagnou NP, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Research. 2004; 32: D537-541.
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009; 25: 1451-1452.
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010; 10: 564-567.
- Lahiry P, Al-Attar SA, Hegele RA. Understanding of β-Thalassemia with focus on the Indian Subcontinent and the Middle East. 2008; 2: 5-10.
- Edison ES, Shaji RV, Devi SG, Moses A, Viswabandhya A, Mathews V, et al. Analysis of beta globin mutations in the Indian population: presence of rare and novel mutations and region-wise heterogeneity. Clin Genet. 2008; 73: 331-337.
- Kumar R, Singh K, Panigrahi I, Agarwal S. Genetic Heterogeneity of Beta Globin Mutations among Asian-Indians and Importance in Genetic Counselling and Diagnosis. Mediterr J Hematol Infect Dis. 2013; 5: e2013003.
- Ibn Ayub M, Moosa MM, Sarwardi G, Khan W, Khan H, Yeasmin S. Mutation analysis of the HBB gene in selected Bangladeshi beta-thalassemic individuals: presence of rare mutations. Genet Test Mol Biomarkers. 2010; 14: 299-302.
- Khan WA, Alam MA, Sarwardi G, Petrou M, Banu B, Hossain B et al. Genotypic study of Bangladeshi thalassemia patients and transfusion requirements. Dhaku Shishu (Child) Hosp J. 1999; 15: 37-39.
- Zahed L. The Spectrum of beta-Thalassemia Mutations in the Arab Populations. J Biomed Biotechnol. 2001; 1: 129-132.
- Treisman R, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned beta-thalassaemia genes. Nature. 1983; 302: 591-596.
- Jamsai D, Zaibak F, Vadolas J, Voullaire L, Fowler KJ, Gazeas S, et al. A humanized BAC transgenic/knockout mouse model for HbE/beta-thalassemia. Genomics. 2006; 88: 309-315.
- Faustino P, Osório-Almeida L, Barbot J, Espírito-Santo D, Gonçalves J, Rom&aTilde;o L, et al. Novel promoter and splice junction defects add to the genetic, clinical or geographic heterogeneity of beta-thalassaemia in the Portuguese population. Hum Genet. 1992; 89: 573-576.
- Agarwal N, Kutlar F, Reading S, Kutlar A, Prchal J. Missense Mutation of the Last Nucleotide of Exon 1 of -globin Gene Interferes with the Expression of Wild Allele. ASH Annual Meeting Abstracts. 2007; 110: 1777.
- Kamel K, el-Najjar A, Webber BB, Chen SS, Wilson JB, Kutlar A, et al. Hb Doha or alpha 2 beta 2[X-N-Met-1(NA1)Val----Glu]; a new beta-chain abnormal hemoglobin observed in a Qatari female. Biochim Biophys Acta. 1985; 831: 257-260
- Harano T, Harano K, Ueda S, Imai K, Ohkuma A, Koya Y, et al. Hb Fukuoka [beta 2(NA2)His----Tyr]: a new mutation at the 2,3-diphosphoglycerate binding site. Hemoglobin. 1990; 14: 199-205.
- Akhavan-Niaki H, Banihashemi A, Azizi M. Beta globin frameworks in thalassemia major patients from north iran. Iran J Pediatr. 2012; 22: 297-302.