
Editorial
Austin J Cancer Clin Res 2014;1(1): 1004.
KRAS – An Evolving Cancer Target
Na Ye and Jia Zhou
Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston
*Corresponding author: Jia Zhou, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, TX 77555, USA
Received: December 20, 2013; Accepted: January 10, 2014; Published: January 12, 2014
The Ras proteins play an important role in cell growth, differentiation, proliferation and survival by regulating diverse cellular pathways. KRAS is the most frequently mutated class of Ras superfamily in all human cancers (21.6%), thus stimulating intensive efforts in developing effective approaches including new drugs to combat various KRAS–mutant–driven cancers. While accumulating evidence supports KRAS as an evolving cancer target, no effective anti–KRAS medications have yet been approved for clinical use to date. Here, we present a concise overview of advances with respect to indirect and direct strategies targeting aberrant KRAS signaling.
The KRAS gene (Ki–ras2 Kirsten RAt Sarcoma viral oncogene homolog), which encoded an approximately 21kDa monomeric, membrane–localized GTPase transforming protein called KRAS, was first discovered and identified as a human oncogene in 1982 [1]. KRAS protein exists as two splice variants, KRAS4A and KRAS4B, all of which belong to the Ras superfamily functioning as molecular switches in regulating diverse cellular pathways for cell growth, differentiation, proliferation and survival. KRAS4B as the dominant isoform is widely expressed in human cells. Ras normally cycles between an active, GTP–bound “off” state and an inactive, GDP–bound “on” state. The conversions are strictly controlled by guanine nucleotide exchange factors (GEFs) and GTPase–activating proteins (GAPs), which are influenced by a number of upstream cell surface receptors such as receptor tyrosine kinases, serpentine receptors, integrins, cytokine receptors and heterotrimeric G–proteins. Activated Ras then targets a number of downstream effectors including PI3K, RAF kinases, MEKK1, Rin 1 and RalGEFs to produce pleiotropic cellular effects. However, the wild–type KRAS gene is a tumor suppressor that can lose this critical function during tumor progression in many types of cancers [2]. Once KRAS mutates, it can become oncogenic and contribute to driving malignant transformation by fixing the protein in a constitutively “on” state. Single point mutations of Ras occur most commonly in residue G12, G13 and Q61, and have been found in 30% of all human tumors. Among these, activating mutations of KRAS are one of the most frequently mutated Ras isoforms in human cancers with the highest prevalence in pancreatic adenocarcinomas (90%), colorectal cancers (45%) and lung cancers (35%) [3].
Based on the important role of Ras in oncogenesis and the prevalence of KRAS mutations in human cancers, KRAS represents an intriguing and promising cancer research target. Over the past decade, KRAS has received a resurgence of interest due to the outcomes of cancer–genomics studies (Figure 1). A number of indirect and direct strategies have been developed to target aberrant KRAS signaling at different levels for inhibiting tumor growth, survival and metastasis. These strategies [4,5] include inhibiting upstream cell surface receptors, inhibiting membrane localization through post–translational modification or trafficking [6], blocking Ras⁄GEFs interactions, enhancing Ras⁄GAP interactions, and directly targeting oncogenic KRAS, as well as inhibiting Ras downstream effectors.
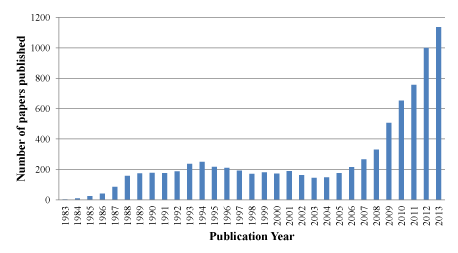
Figure 1: Number of papers published between 1983 and 2013 according to recent Pubmed search using “KRAS”.
Several of the new therapeutic agents as indirect treatments have shown promising results in human clinical trials leading to the development of many additional indirect strategies. Farnesyltransferase inhibitors (FTIs), which inhibit the posttranslational modification of Ras proteins by FTase, have advanced into human clinical trials for cancer treatment, either alone or in combination with conventional cytotoxic drugs [7]. Of these, two peptidomimetics, Tipifarnib (R115777) and Lonafarnib (SCH66336), are undergoing the most significant clinical evaluation. In addition, the farnesyl isoprenoid–containing small molecule Salirasib, which disrupts the association of chronically active Ras proteins with the plasma membrane thereby blocking the function of all Ras isoforms, is well tolerated in patients with KRAS mutated lung adenocarcinomas from phase II clinical trial study report [8]. Another important strategy in the treatment of KRAS mutant cancers is to exploit small molecule inhibitors targeting upstream cell surface receptors, including epidermal growth factor receptor (EGFR), and downstream Ras effector signaling pathways, including the Raf–MEK–ERK, PI3K–AKT–mTOR and GEF–Ral pathways. Cetuximab, an EGFR inhibitor, is in phase II clinical trial assessment as monotherapy for the treatment of KRAS mutated refractory colorectal cancer (NCT01450319) [9]. Trametinib (GSK1120212, JTP–74057), an orally bioavailable, tetrahydropyridopyrimidine–based, highly specific and potent allosteric inhibitor of MEK1 and MEK2, is currently in phase II study as the second line treatment of stage IV KRAS–mutant nonsmall cell lung cancer patients (NCT01362296) [9].
Nevertheless, the mutant KRAS related treatments have proven challenging in the clinic. In FTI–treated cells, KRAS can also be subject to alternative prenylation by geranylgeranyltransferase type I (GGTase I) [10], followed by continued membrane localization of KRAS and concomitant upregulation of downstream signaling, thereby leading to very poor efficacy of FTI monotherapy in clinical trials. Additionally, EGFR inhibitors, Gefitinib and erlotinib, show significant effects on blocking KRAS signaling, but their effects are very short due to the rapid development of drug resistance [4]. Moreover, due to the existence of negative feedback loops on the reciprocal pathway, Ras mutant cancer cells show limited response to single–agent pathway inhibitors leading to no significant effect on suppressing tumor growth [11]. Currently, drug combinatorial therapies that target multiple points within the Ras signaling network are believed to be one of the most promising directions and several approaches benefitting from this strategy have advanced into the human clinical trials, including FTI inhibitors with cytotoxic or hormonal agents, concurrent inhibition of the Raf–MEK–ERK & PI3K–AKT–mTOR pathways, inhibition of the Raf–MEK–ERK MAPK & PI3K–AKT–mTOR pathways with chemotherapy or radiotherapy. Phase II clinical trials have shown that Lonafarnib in combination with Gemcitabine (a nucleoside analog used as chemotherapy) displays a promising response rate as a second line treatment in advanced metastatic urothelial cancer (NCT00006351) [9]. Sorafenib, a Raf–1 inhibitor, in combination with a topoisomerase 1 inhibitor, Irinotecan, is currently in Phase II clinical trial assessment in patients with KRAS mutated metastatic colorectal tumors after failure of all drugs known to be effective (NCT01715441) [9].
Despite the significant progress achieved over the years in anti–KRAS drug development involving indirect approaches, no effective direct pharmacological inhibitor of the Ras oncoprotein has reached the clinic to date. For years, KRAS has been considered as an “undruggable” target, because numerous efforts made by industry and academic laboratories have failed to design a drug to directly target KRAS. The cause of these failures derives from the conventional conception that KRAS inhibitors are supposed to successfully block KRAS signaling by directly targeting its GTP binding pocket. However, this turns out to be very difficult and challenging with the following possible reasons: 1) the role of GDP or GTP is to stabilize “on” or“off” states of the Ras protein, rather than as a substrate of catalytic reactions; 2) the picomolar affinity between KRAS and GTP; 3) the micromolar concentration of GTP in the cell; and 4) KRAS activation and signaling accomplished through protein–protein interactions (PPIs) with GEFs, GAPs and various KRAS effector proteins, etc. To overcome such challenges, the US National Cancer Institute (NCI) this year allocated US$10 million specifically to develop novel ideas, new drugs or therapies for blocking Ras. In fact, such efforts toward direct inhibition of Ras have started to yield promising initial results [12– 15]. With the combined analysis of computational and cell biology, Hancock and Gorfe [16] have demonstrated that andrographolide, a bicyclic diterpenoid lactone isolated from Andrographis paniculata, and its benzylidene derivatives can block GDP–GTP exchange and inhibit both wild–type and oncogenic KRAS signaling by directly binding to the switch regions of Ras, preferentially targeting a transient pore behind switch 1 as well as a groove between switches 1 and 2. This finding suggests that inhibiting nucleotide exchange is a valid approach to abrogating the function of oncogenic mutant Ras. Very recently, Shokat and his team [17], for the first time, developed an innovative approach for specifically targeting a type of KRAS mutant called G12⁄C (for Glycine–12 to Cysteine), which occurred in 2% of all human cancers. Taking advantage of the nucleophilicity of the cysteine thiol (SH) group in the KRAS (G12C) mutation, they first developed direct KRAS inhibitors by using a disulfide–fragmentbased screening approach called tethering, followed by optimizing their prototype compounds to obtain novel acrylamides and vinyl sulfonamides. These molecules can irreversibly form stable cysteine bond to avoid rapid degradation of disulfide (S–S) bond and prefer to bind the GDP–bound form of Ras. Meanwhile, with the obtained X–ray crystal structure of an inhibitor–tethered protein, they have also identified an undiscovered allosteric pocket, S–IIP, presenting new opportunities for drug development [17]. More recently, Gray and his team disclosed a GDP analogue, SML–8–73–1, and its prodrug derivative, SML–10–70–1, which are identified as selective, directacting covalent inhibitors of the KRAS G12C mutant relative to wild–type Ras[18]. SML–8–73–1 can access the active site of mutated KRAS more directly than the Shokat inhibitors [17], but it must be administered as a cell–permeable precursor [18].
While substantial advances in understanding Ras biology and functions over the past thirty years have been made, all indirect and direct strategies targeting aberrant KRAS signaling are still in early exploratory stage and none of them has yet reached the market. More extensive efforts are essential to fulfill clinical expectations, especially by developing novel approaches to attacking KRAS protein directly. Other than blocking the main KRAS signaling pathways that we have discussed above, the inhibition of novel relevant targets is also a good choice for anti–KRAS, for instance, the cell cycle related kinase PLK1 [19]. In addition, the further development and application of high–throughput genome–wide unbiased functional screening efforts to block Ras expression, such as the therapeutic use of RNA interference technology [11] to search for synthetic lethal partners of mutant KRAS and novel methods for the targeted delivery of siRNA to the tumor [20], will also likely yield novel, unexpected and more tractable directions for the discovery of anti–Ras inhibitors. While the urgent need for breakthroughs in the field is evident, the continued investigation into this promising cancer target will pave the way toward new and effective therapies that can benefit patients with KRAS–mutant–driven cancers in the years to come.
Acknowledgments
This work was supported by Grants P50 CA097007, P30 DA028821, and R21 MH093844 from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at the University of Texas Medical Branch (UTMB).
References
- Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A. 1982; 79: 3637-3640.
- Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001; 29: 25-33.
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3: 11-22.
- Wang Y, Kaiser CE, Frett B, Li HY. Targeting Mutant KRAS for Anticancer Therapeutics: A Review of Novel Small Molecule Modulators. J Med Chem. 2013; 56: 5219-5230.
- Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005; 1756: 127-144.
- van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013; 33: 237-251.
- Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006; 47: 15-31.
- Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011; 6: 1435-1437.
- https://www.clinicaltrials.gov/
- Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011; 3: 1787-1808.
- Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011; 2: 359-372.
- Grant BJ, Lukman S, Hocker HJ, Sayyah J, Brown JH. Novel allosteric sites on Ras for lead generation. PLoS One. 2011; 6: e25711.
- Buhrman G, O'Connor C, Zerbe B, Kearney BM, Napoleon R. Analysis of binding site hot spots on the surface of Ras GTPase. J Mol Biol. 2011; 413: 773-789.
- Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012; 109: 5299-5304.
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012; 51: 6140-6143.
- Hocker HJ, Cho KJ, Chen CY, Rambahal N, Sagineedu SR. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc Natl Acad Sci U S A. 2013; 110: 10201-10206.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013; 503: 548-551.
- Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG. Therapeutic targeting of oncogenic k-ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 2014; 53: 199-204.
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009; 137: 835-848.
- Zeng L, Li J, Wang Y, Qian C, Chen Y, et al. Combination of siRNA-directed KRAS oncogene silencing and arsenic-induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine. 2013