
Review Article
Austin J Cardiovasc Dis Atherosclerosis. 2016; 3(1): 1017.
Vascular Inflammation and Hypertension
Ogura S1,2,3*, Latapati R2, Shimosawa T2 and Nakayama T1,4
1Division of Laboratory Medicine, Department of Pathology and Microbiology, Nihon University School of Medicine, Tokyo, Japan
2Department of Clinical Laboratory, The University of Tokyo, Tokyo, Japan
3Division of Clinical Epigenetics, Research Center for Advanced Science and Technology, The University of Tokyo, Japan
4Division of Companion Diagnostics, Department of Pathology of Microbiology, Nihon University School of Medicine, Tokyo, Japan
*Corresponding author: Sayoko Ogura, Division of Laboratory Medicine, Department of Pathology and Microbiology, Nihon University School of Medicine, 30-1 Ooyaguchi-kamimachi, Itabashi-ku, Tokyo, Japan
Received: March 18, 2016; Accepted: April 04, 2016; Published: April 06, 2016
Abstract
Hypertension is a very common disease and is often associated with left ventricular hypertrophy, obesity, diabetes and dyslipidemia. The consequences of hypertension involve coronary heart disease, heart failure, renal failure, stroke and exacerbation of the occlusive atherosclerotic coronary arterial disease. These disease states are associated with vascular structural and functional inflammatory changes including endothelial dysfunction, altered vasomotor tone, and vascular remodeling. However, whether vascular inflammation is a cause or result of hypertension is not well understood. Vascular inflammation and hypertension may share somecommon pathophysiological mechanism. In this review will show recent data concerning a potential link between inflammation and hypertension, including CRP, oxidative stress, RAS, prostaglandin, adaptive immune system, and Th17 activation by high salt intake, a major risk factor for developing hypertension.
Keywords: Oxidative stress; LOX-1; Adrenomedullin; Aldosterone; COX-2; Th17
Introduction
Hypertension is a major cause of morbidity and mortality worldwide. Over the past years, a plethora of information has established the diagnostic and prognostic value of various mediators of vascular inflammation in hypertension. However it is difficult to explain clearly what causes hypertension in human study. Many animal or in vitro models explain on the relation vascular inflammation and hypertension. This review focuses on the relation vascular inflammation and hypertension by the possible mediators, CRP (C-reactive protein), adaptive immune response, oxidative stress, RAS (renin-angiotensin-aldosterone system), prostaglandin, and Th17 activation by high salt intake (Figure 1).
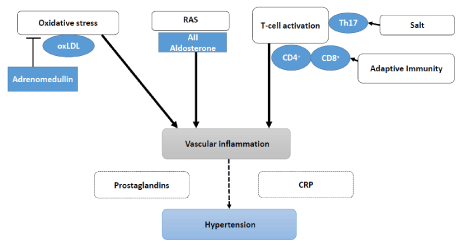
Figure 1: Schematic diagram illustrating the relationship between vascular inflammation and hypertension. Working hypothesis describing hypertensive stimuli (white circle) induces vascular inflammation. RAS: renin-angiotensin-aldosterone system.
CRP (C - Reactive Protein)
CRP is considered the inflammatory marker with the strongest association with hypertension. It has been demonstrated in numerous clinical trials that hypertensive patients commonly have increased plasma CRP levels [1]. Nonhypertensive offspring of hypertensive parents tend to have higher serum CRP levels than offspring of nonhypertensive patients [2]. Elevated HS-CRP is both a risk marker and risk factor for hypertension and cardiovascular disease [3]. Increases in HS-CRP (over 3μg/mL) may increase blood pressure in just a few days in dose dependent fashion [4]. Increases in high sensitivity CRP (HS-CRP) as well as other inflammatory cytokines such as interleukin-1B, (IL-1B), IL-6, tumor necrosis alpha (TNF-a) and chronic leukocytosis are observed in hypertension and hypertension-related target organ damages, such as increased carotid intima media thickness (IMT) [5]. CRP is an acute phase protein, but it also can stimulate monocytes to release proinflammatory cytokines such as interleukin-6 (IL-6), interleukin-1 beta (IL-1β), and tumor necrosis factor alpha (TNF-a) and also acts on endothelial cells to express intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, these effects further promote inflammation. CRP is only marker for inflammation, what stimuli increases CRP and whether vascular inflammation increase CRP are not known.
Oxidative Stress
In human hypertension, biomarkers of systemic oxidative stress are elevated [6,7]. CRP levels also have been shown to correlate with the level of oxidative stress in inflammatory cells from hypertensive patients [8,9]. Nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase is a major source of ROS in immune cells and also in the vasculature. Excessive ROS levels can also induce cellular damage by interacting with DNA, lipids, and proteins, which may further impair vascular structure and function [10]. Reactive oxygen species (ROS) is defined as oxygen with unpaired electron and highly active chemical. Superoxide anion (O2 -), hydroxyl radical (HO♦), hydrogen peroxide (H2O2), peroxynirtite (ONOO-) and lipid radical are classified ROS [11]. In cellular matrix or membrane, in addition to NADPH oxidase, xanthine oxidase, cyclooxygenase, lipoxygenase, NO synthase, hemeoxygenase, peroxygenase, or heme protein are potential enzymes to produce ROS. In vascular endothelial cells, eNOS which is a cytochrome P450 reductase-like enzyme is important source for ROS. eNOS utilizes tetrabiopterin (BH4) to produce NO from L-arginine and under deficiency of L-arginine or BH4, eNOS produces O2 - or H2O2. Physiologically, ROSs are produced in a controlled manner at low concentrations and function as signaling molecules to maintain vascular integrity by regulating endothelial function and vascular contraction-relaxation balance. ROS activates Ca2+ signal, tyrosine kinases or mitogen-activated protein kinases (MAPK) by non-genomic action and increases expression of MCP- 1, VCAM-1, ICAM-1 and atherogenic genes by genomic action via activating NF-κB. Under pathological conditions, increased ROS bioactivity leads to vascular inflammation which isan essential pathophysiological mechanism in the development of hypertension.
Oxidized LDL
Oxidized LDL is produced by oxidation of low density lipoprotein (LDL) and its oxidation is caused by peroxyradical and radical chain reactions. Source of ROS are leukocytes, macrophage, endothelial cells as well as vascular smooth muscle cells. Oxidized LDL increases MCP-1 secretion from endothelial cells and further induces monocyte and macrophage infiltration into vasculature [12]. Lectin-like oxidized low-density lipoprotein receptor (LOX-1) mRNA expression is minimal in the aorta from normotensive rats, but is markedly up-regulated in spontaneously hypertensive rats and salt-loaded Dahl salt-sensitive rats, suggesting a correlation between LOX-1 and hypertension [13]. However, there has been little information on the relationship between LOX-1 and hypertension in humans. In a study of healthy workers, serum LOX-1 ligand containing ApoB (LAB) level was correlated with only diastolic but not systolic blood pressure [14]. LOX-1 is also regulated by oxidative stress [15,16], and oxidized LDL induces ROS via LOX-1, therefore there are vicious cycles among ROS [17]. Cellular and organ damages by Oxidized LDL-induced ROS are not limited in endothelial cells but cardiomyocyte remodeling after ischemia or inflammation and fibrosis in the kidney [12].
Adrenomedullin
The endogenous and exogenous antioxidants that have demonstrated an ability to alter the function of blood vessels and participate in the main redox reactions involved in vascular inflammation. Adrenomedullin (AM) was identified by Kitamura in 1993 as a potent vasodilating peptide [18] and the studies using its deficient mice model revealed that AM is a potent intrinsic antioxidant. Plasma adrenomedullin concentrations are elevated in many hypertensive patients [19]. In AM deficient mice, overall ROS marker, 8-isoprostaglandin F2 excretion is high and angiotensin II plus salt loading induced local ROS production in the heart and marked pericoronary fibrosis and narrowing independent from blood pressure [20]. In other model, cuff-induced vascular damage was reduced by topical administration of AM via viral vector and this effect was in parallel with reduction of ROS [21]. These models are closely related local or systemic renin-angiotensin system which is a strong inducer of NADPH oxidase and oxidative stress. In vitro experiments showed that AM interfere with angiotensin II signaling and inhibits NADPH oxidase activity [22].
Antioxidants and hypertension
Antioxidants for hypertension are the study of, in which a peptide inhibitor of the NADPH oxidase was shown to lower blood pressure and to prevent macrophage accumulation [23]. Roson et al showed that the acute infusion of sodium caused an increase in renal levels of chemokine ligand 5 (RANTES), NFκB, HIF1a, and angiotensin II in rats, and that superoxide dismutase (SOD) mimetic, Tempol, markedly reduced these responses [24].The above mentioned intrinsic antioxidants are scavengers and also exogenous antioxidants are studied for long time in human. Vitamin C is a potent water-soluble antioxidant. On the vascular wall behaves as enzyme modulator exerting up-regulation on eNOS and down regulation of NOX [25] Nevertheless, there are several clinical trials in which the effect of vitamin C supplements on blood pressure have yielded inconsistent findings [26]. Recently cholesterol lowering drugs, probucol and statins are reported to have antioxidant property as their pleiotropic effects [27]. However, the dietary intake of antioxidants and polyphenols could have an effect in the primary prevention or reduction of hypertension. [28]. There are some reports that those exogenous ROS scavengers are effective in preventing hypertension, but there remain controversial reports [29].
RAS (Renin-Angiotensin System)
Angiotensin II and Aldosterone are well known to the reninangiotensin system (RAS), they are strongly inducer for vascular inflammation. RAS plays a crucial role in the initiation and maintenance of vascular inflammation and vascular remodeling [30]. Vascular inflammation leads to endothelium dysfunction. A dysfunctional endothelium is leaky and facilitates migration of inflammatory cell into the vascular wall and stimulates smooth muscle cells proliferation.
Angiotensin II
More convincing support has been provided by the use of ACE inhibitors in recent clinical trials in which profound effects of ACE inhibitors on reducing cardiovascular events were seen. Angiotensin receptor blockers (ARBs) may also reduce inflammation not only decrease blood pressure [31]. In animal model, acute treatment with Ang II significantly increases inflammatory changes, leukocytes adhesion in mesenteric arteries [32]. Moreover, animal and human studies show that Ang II induces proinflammatory responses in arteries, heart, and kidney by regulating the expression of cytokines and chemokines. Macrophages are components of the innate immune system, and are poised to respond to non-specific stimuli, such as might be present in the tissue damage induced by angiotensin II [33]. It is also now well-established that Ang II activates NADPH oxidases in VSMCs, monocytes, macrophages, and endothelial cells to produce reactive oxidant species [34]. In the setting of Ang IIinduced hypertension, higher levels of subunit of NADHP oxidase expressions, including p47phox, p22phox, and NOX2, components of NOX2 oxidase. Furthermore, adoptive transfer of T cells deficient in NADPH oxidase results in lower superoxide production and blood pressurein response to Ang II [35].
Aldosterone
Clinical studies indicate that the prevalence of hyperaldosteronism may be increased in resistant hypertension, that aldosterone concentrations “escape” to pretreatment levels during chronic treatment of congestive heart failure or hypertension with an Ang-converting enzyme (ACE) inhibitor or Ang receptor blocker (ARB) [36]. Seminal studies in rat models demonstrated that MR activation causes perivascular and interstitial fibrosis [37]. Rocha et al., demonstrated that treatment with aldosterone and salt caused extensive inflammatory arterial lesions with perivascular macrophages in the heart [38]. MR antagonism decreases aortic inflammation, fibrosis,and hypertrophy in hypertensive rats [39]. However, the heart and the vasculature do not have enough expression of 11β hydroxysteroid dehydrogenase type II, there are some reports the genomic activation by the mechanism of Aldo binds MR and expresses its pro-inflammatory action without glucocorticoid deactivation [40]. Despite lowering plasma aldosterone, salt worsens renal injury by paradoxical activation of the mineralocorticoid receptor (MR) [41]. Fujita et al showed two pathways involving aldosterone-MR and renal SNS-GR that contribute to an impaired capacity to excrete sodium [42-44].
Like Ang II, aldosterone activates NADPH oxidases in rat VSMCs [45]. Increased oxidative stress activates the redox sensitive NF-κB, and triggers inflammation. Hence, aldosterone-stimulated activation of vascular inflammation by oxidative stress and NF-κB. Besides rac1, a component of NADPH oxidase, can translocate mineralocorticoid receptor into nucleus independent from aldosterone and exerts its genomic effect to induce its target gene transcription such as sgk1 [42]. This indicates that oxidative stress can activate mineralocorticoid receptor in organs even aldosterone level is low [46,47]. It implies that mineralocorticoid receptor blockade and reduction of salt intake are possibly effective in reducing inflammation and preserving vascular function in hypertensive patients [48].
Prostaglandins
A number of animal studies and observations in human hypertensive subjects suggest that the prostaglandin system plays a role in the pathogenesis of hypertension. Inflammation activates phospholipase A2 (PLA2) to release Arachidonic Acid (AA), whose metabolism by cyclooxygenases (COXs) generates prostaglandins (PGs). PGs normally have an antihypertensive action. Prostacyclin (PGI2) inhibits platelet aggregation and vasoconstriction. PGI2 synthase (PTGIS), a catalyst of PGI2 synthesis from prostaglandin H2, is widely distributed and predominantly found in vascular endothelial and smooth muscle cells. However, PGH2 (prostaglandin endoperoxide), thromboxane (Tx)A2 generated by TxA2 synthase (TxA2-2), and isoprostanes (Iso) can constrict blood vessels [49] (Figure 2). Recent animal studies have shown that PGI2 may, in fact, paradoxically induce vasoconstriction rather than vasodilatation in certain circumstances. In the aortic rings from SHR and aged Wistar Kyoto rats, the endothelium dependent contractions elicited by acetylcholine most likely involve the release of PGI2 with a concomitant contribution of PGH2 [50] In previous studies, mice deficient in the prostaglandin E2 (PGE2) EP2 receptor is low in resting systolic blood pressure (BP) than that of wild-type controls [51]. The BP of those mice increased blood pressure when they were put on a high-salt diet, suggesting that the EP2 receptor is involved in sodium handling inthe kidney [52]. We analyzed three single-nucleotide polymorphisms (SNP) in the human PTGER2 gene the A/A type of the SNP rs17197 (rs17197, A/G in 3’UTR) was significantly more frequent in EH subjects than in NT subjects [53]. Synthesis of PGI2 is enhanced in the spontaneously hypertensive and Gold blat hypertensive rat [54]. We and other group identified several mutations in the human PGI2 synthase (PTGIS) gene in a family with a history of essential hypertension, but haplotype of PTGIS genes were not associated in essential hypertension [55-62]. Metabolism of PGE2, PGF2-a, and PGI2 by prostaglandin 15-hydroxydehydrogenase is impaired in hypertensive genetic models [63]. In either case, prostanoids cause both prohypertensive and antihypertensive effects by acting on blood vessels.
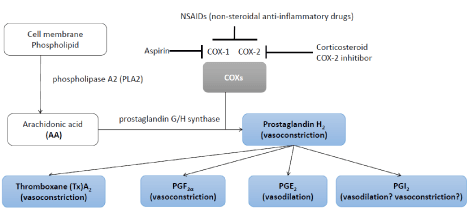
Figure 2: Schematic diagram illustrating synthesis and metabolism of Eicosanoid. The prostaglandins (PG), thromboxanes (TX), and lipoxygenase and
epoxygenase products are collectively called eicosanoids. Arachinoid Acid (AA) is released from cell membrane phospholipid by phospholipases (PLAs) and metabolized Prostagrandin H2 by the sequential actions of prostaglandin G/H synthase, or cyclooxygenase (COX), and respective synthases. Anti-inflammatory
drugs (Asprin, NSAID, Corticosteroid, COX-2 inhibitor) inhibit COXs (COX-1,2) activation.
COX inhibitors
Cyclooxygenase (COX) catalyzes the synthesis of prostaglandins (PGs) from arachidonate. The cyclooxygenase-derived prostanoids (e.g., PGE2, PGF2_, PGD2, PGI2, thromboxane A2) are generated by either the largely constitutive isoform COX-1 or by the inducible isoform COX-2. Two isozymes encoded by different genes, cox1and cox2, mediate this process. Accumulating evidence indicates COX-1 and COX-2 activity differentially influence renal and cardiovascular function. For example patients receiving a selective COX2 inhibitor exhibited an increased incidence of thrombotic cardiovascular events and hypertension [64]. In addition to the well-recognized gastrointestinal toxicity caused by nonspecific COX inhibitor (NSAIDs), these agents have also been found to produce a mean increase in blood pressure of 5.0mm Hg [65]. In contrast, low-dose aspirin, which primarily inhibits COX1, can lower blood pressure in healthy subjects with mild hypertension, [67,68]. Future studies are needed to assess the effect.
Adaptive Immune Response
Several recent investigations have further defined the role of immune system. Particularly the adaptive immune system, in hypertension, provided novel insights into the genesis of hypertension, and identified novel targets for the treatment of hypertension [69]. The first line of defense against pathogens is the innate immune response. In contrast to the innate immune system, the adaptive immune system is highly specific. Grollman et al., showed that immunosuppression attenuates hypertension in rats [70]. Ba et al. found that transplanting the thymus from a Wistar- Kyoto (WKY) rat to a spontaneously hypertensive rat (SHR) resulted in a decrease in blood pressure in the SHR [71]. Based upon this finding, more in precise mechanism how immune responses by either T and B cells regulate blood pressure has been studied. Mice lacking recombinase-activating gene 1 (Rag-1_/_ mice) cannot generate functional T cell receptors or B cell antibodies and thus lack both T and B lymphocytes [35]. The increase in blood pressure caused by either Ang II or DOCA salt was significantly blunted in Rag-1_/_ mice, suggesting that either T or B cells mediate overt hypertension. Rag-1_/_ mice did not exhibit increased vascular superoxide production and endothelial dysfunction. This results shows that T cells play a major role in hypertension. Several recent studies suggest a vascular protective effect of T regulatory cells, much in the same way that these cells may provide renal protection [72]. An association between vascular inflammation and T regulatory cells was initially described in salt-sensitive hypertension by comparing vascular inflammatory markers and T cells in Dahl rats with chromosome 2 from the Brown Norway rat in which regulatory T cell function is suppressed by increased FOXP3. The congenic rats exhibited reduced vascular inflammation and increased vascular expression of Foxp3, a transcription factor specific to T regulatory cells [73]. In other study, either mineralocorticoid or AngII-dependent hypertension model, adoptive transfer of T regulatory cells blunts the hypertension and prevents the development of impaired mesenteric artery function and remodeling [74]. Immune cell in the conduit vessels might be an important factor for what is occurring in resistance vessels, especially the renal microvasculature, as a mechanism to promote the development of hypertension.
T Cell Activation by Salt
The relevance of the renal inflammation in the pathogenesis of Salt Sensitive Hypertension (SSHTN) is underlined by the demonstration that treatments that suppress the renal inflammation result in amelioration or prevention of salt-driven hypertension. The connection between sodium intake and health is manifested by the relationship between sodium intake and blood pressure. Several studies revealed salt sensitive animal models showed inflammation in the kidney and changed inflammatory gene expressions [75]. Saltsensitive hypertension with increased renal inflammation as a result of T cell imbalance, dysregulation of CD4+and CD8+lymphocytes and chronic leukocytosis with increased neutrophils and reduced lymphocytes [76].
Patients with hypertensive nephrosclerosis have higher renal infiltration of CD4+ and CD8+ T cells than normotensive control patients. Furthermore, circulating levels of chemokines have been reported to be elevated in hypertensive patients [77]. Pro-inflammatory cytokine Interlaukin-17 (IL-17) contributes hypertension [78]. IL- 17 is produced by Th17cells.Two recent studies collectively suggest that excess sodium drives autoimmunity at the cellular level [79,80]. One group of researchers had showed increased Th17 cell numbers in the blood of people who consumed high salt diet, they conducted experiments on the effects of elevated sodium concentrations on the differentiation of immature human T cells into pathogenic Th17 cells. They indeed found that high sodium concentrations drove a dramatic increase in differentiation into pathogenic Th17 cells in vitro. To strengthen these findings, they fed mice predisposed to a Th17-related autoimmune disease either a standard or high-salt diet. The high-salt diet accelerated the development of the autoimmune disease, and the symptoms were more severe on the high-salt diet than on the standard diet.
Conclusion
The recent studies have shown the relevance of inflammation and hypertension, including its mediators. C-Reactive Protein (CRP) is considered the inflammatory marker with the strongest association with hypertension. Oxidative stress, RAS are known to be associated with inflammation and can contribute to hypertension. T-cells activation at least in part elevate blood pressure by exacerbating autoimmune response in vasculature and possibly in the kidney. However, some anti-inflammatory drugs (NSAID or COX-2 inhibitor) paradoxically cause hypertension in human. Because immunosuppressants can have serious side effect (such as sodium retention, inhibit angiogenesis).Clinical studies investigating antioxidant supplements have failed to show any consistent benefit. Most clinical studies on anti-oxidants were not enough to scavenge oxidative stress and it also become free radical in body, if there were pro-oxidant conversion. New anti-inflammatory drugs could be used for prevent hypertension, or vascular inflammation in future.
Sources of Funding
This work was supported in part by Grants-in-Aid from the Japan Society for the Promotion of Science (JSPS) fellows (26461262).
References
- Blake GJ, Rifai N, Buring JE, Ridker PM. Blood pressure, C-reactive protein, and risk of future cardiovascular events. Circulation. 2003; 108: 2993-2999.
- Lieb W, Pencina MJ, Wang TJ, Larson MG, Lanier KJ, Benjamin EJ, et al. Association of parental hypertension with concentrations of select biomarkers in nonhypertensive offspring. Hypertension. 2008; 52: 381-386.
- Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, et al. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. 2007; 115: 1020-1028.
- Razzouk L, Muntner P, Bansilal S, Kini AS, Aneja A, Mozes J, et al. C-reactive protein predicts long-term mortality independently of low-density lipoprotein cholesterol in patients undergoing percutaneous coronary intervention. Am Heart J. 2009; 158: 277-283.
- Ghanem FA, Movahed A. Inflammation in high blood pressure: a clinician perspective. J Am Soc Hypertens. 2007; 1: 113-119.
- Grossman E. Does increased oxidative stress cause hypertension? Diabetes Care. 2008; 31 Suppl 2: S185-189.
- Rodrigo R, González J, Paoletto F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens Res. 2011; 34: 431-440.
- Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal. 2014; 20: 102-120.
- Al-Delaimy WK, Jansen EH, Peeters PH, van der Laan JD, van Noord PA, Boshuizen HC, et al. Reliability of biomarkers of iron status, blood lipids, oxidative stress, vitamin d, c-reactive protein and fructosamine in two dutch cohorts. Biomarkers. 2006; 11: 370-382.
- Hassoun EA, Li F, Abushaban A, Stohs SJ. Production of superoxide anion, lipid peroxidation and DNA damage in the hepatic and brain tissues of rats after subchronic exposure to mixtures of TCDD and its congeners. J Appl Toxicol. 2001; 21: 211-219.
- Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003; 108: 1912-1916.
- Ogura S, Kakino A, Sato Y, Fujita Y, Iwamoto S, Otsui K, et al. Lox-1: the multifunctional receptor underlying cardiovascular dysfunction. Circ J. 2009; 73: 1993-1999.
- Nagase M, Kaname S, Nagase T, Wang G, Ando K, Sawamura T, et al. Expression of LOX-1, an oxidized low-density lipoprotein receptor, in experimental hypertensive glomerulosclerosis. J Am Soc Nephrol. 2000; 11: 1826-1836.
- Sawamura T, Wakabayashi I, Okamura T. LOX-1 in atherosclerotic disease. Clin Chim Acta. 2015; 440: 157-163.
- Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997; 386: 73-77.
- Nagase M, Ando K, Nagase T, Kaname S, Sawamura T, Fujita T. Redox-sensitive regulation of lox-1 gene expression in vascular endothelium. Biochem Biophys Res Commun. 2001; 281: 720-725.
- Ogura S, Shimosawa T, Mu S, Sonobe T, Kawakami-Mori F, Wang H, et al. Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized ldl receptor.American journal of physiology Heart and circulatory physiology. 2013; 305: H155-162.
- Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, et al. Adrenomedullin: A novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993; 192: 553-560.
- Kohno M, Hanehira T, Kano H, Horio T, Yokokawa K, Ikeda M, et al. Plasma adrenomedullin concentrations in essential hypertension. Hypertension. 1996; 27: 102-107.
- Shimosawa T, Shibagaki Y, Ishibashi K, Kitamura K, Kangawa K, Kato S, et al. Adrenomedullin, an endogenous peptide, counteracts cardiovascular damage. Circulation. 2002; 105: 106-111.
- Kawai J, Ando K, Tojo A, Shimosawa T, Takahashi K, Onozato ML, et al. Endogenous adrenomedullin protects against vascular response to injury in mice. Circulation. 2004; 109: 1147-1153.
- Liu J, Shimosawa T, Matsui H, Meng F, Supowit SC, DiPette DJ, et al. Adrenomedullin inhibits angiotensin II-induced oxidative stress via Csk-mediated inhibition of Src activity. Am J Physiol Heart Circ Physiol. 2007; 292: H1714-1721.
- Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003; 23: 776-782.
- Roson MI, Della Penna SL, Cao G, Gorzalczany S, Pandolfo M, et al. Different protective actions of losartan and tempol on the renal inflammatory response to acute sodium overload. J Cell Physiol. 2010; 224: 41-48.
- Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension. 2003; 41: 534-539.
- Block G, Mangels AR, Norkus EP, Patterson BH, Levander OA, Taylor PR. Ascorbic acid status and subsequent diastolic and systolic blood pressure. Hypertension. 2001; 37: 261-267.
- Hermida N, Balligand JL. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: The role of statins. Antioxid Redox Signal. 2014; 20: 1216-1237.
- Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, et al. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr. 2003; 22: 18-35.
- González J, Valls N, Brito R, Rodrigo R. Essential hypertension and oxidative stress: New insights. World J Cardiol. 2014; 6: 353-366.
- Renna NF, Lembo C, Diez E, Miatello RM. Role of Renin-Angiotensin system and oxidative stress on vascular inflammation in insulin resistence model. Int J Hypertens. 2013; 2013: 420979.
- Mansur SJ, Hage FG, Oparil S. Have the renin-angiotensin-aldosterone system perturbations in cardiovascular disease been exhausted? Curr Cardiol Rep. 2010; 12: 450-463.
- Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, et al. Direct evidence of leukocyte adhesion in arterioles by angiotensin ii. Blood. 2004; 104: 402-408.
- Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012; 3: 128.
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994; 74: 1141-1148.
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007; 204: 2449-2460.
- Sato A, Saruta T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. J Int Med Res. 2001; 29: 13-21.
- Brown NJ. Aldosterone and vascular inflammation. Hypertension. 2008; 51: 161-167.
- Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002; 283: H1802-1810.
- Benetos A, Lacolley P, Safar ME. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 1997; 17: 1152-1156.
- Kiyosue A, Nagata D, Myojo M, Sato T, Takahashi M, Satonaka H, et al. Aldosterone-induced osteopontin gene transcription in vascular smooth muscle cells involves glucocorticoid response element. Hypertens Res. 2011; 34: 1283-1287.
- Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: Role of oxidative stress. Hypertension. 2007; 50: 877-883.
- Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, et al. Rac1 gtpase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. The Journal of clinical investigation. 2011; 121: 3233-3243.
- Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, et al. Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011; 17: 573-580.
- Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol. 2014; 25: 1148-1155.
- Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension. 2005; 45: 773-779.
- Wang H, Shimosawa T, Matsui H, Kaneko T, Ogura S, et al. Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens. 2008; 26: 1453-1462.
- Nagase M, Ayuzawa N, Kawarazaki W, Ishizawa K, Ueda K, Yoshida S, et al. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. Hypertension. 2012; 59: 500-506.
- Ogura S, Shimosawa T. Oxidative stress and organ damages. Curr Hypertens Rep. 2014; 16: 452.
- Lin L, Mistry M, Stier CT Jr, Nasjletti A. Role of prostanoids in renin-dependent and renin-independent hypertension. Hypertension. 1991; 17: 517-525.
- Gluais P, Paysant J, Badier-Commander C, Verbeuren T, Vanhoutte PM, Feletou M. In shr aorta, calcium ionophore a-23187 releases prostacyclin and thromboxane a2 as endothelium-derived contracting factors. Am J Physiol Heart Circ Physiol. 2006; 291: H2255-2264.
- Tilley SL, Audoly LP, Hicks EH, Kim HS, Flannery PJ, Coffman TM, et al. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999; 103: 1539-1545.
- Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999; 5: 217-220.
- Sato M, Nakayama T, Soma M, Aoi N, Kosuge K, Haketa A, et al. Association between prostaglandin e2 receptor gene and essential hypertension. Prostaglandins Leukot Essent Fatty Acids. 2007; 77: 15-20.
- Welch WJ, Patel K, Modlinger P, Mendonca M, Kawada N, Dennehy K, et al. Roles of vasoconstrictor prostaglandins, COX-1 and -2, and AT1, AT2, and TP receptors in a rat model of early 2K,1C hypertension. Am J Physiol Heart Circ Physiol. 2007; 293: H2644-2649.
- Iwai N, Katsuya T, Ishikawa K, Mannami T, Ogata J, Higaki J, et al. Human prostacyclin synthase gene and hypertension : the Suita Study. Circulation. 1999; 100: 2231-2236.
- Nakayama T. Genetic polymorphisms of prostacyclin synthase gene and cardiovascular disease. Int Angiol. 2010; 29: 33-42.
- Nakayama T, Soma M, Haketa A, Aoi N, Kosuge K, Sato M, et al. Haplotype analysis of the prostacyclin synthase gene and essential hypertension. Hypertens Res. 2003; 26: 553-557.
- Nakayama T, Soma M, Kanmatsuse K. Organization of the human prostacyclin synthase gene and association analysis of a novel CA repeat in essential hypertension. Adv Exp Med Biol. 1997; 433: 127-130.
- Nakayama T, Soma M, Rahmutula D, Tobe H, Sato M, Uwabo J, et al. Association study between a novel single nucleotide polymorphism of the promoter region of the prostacyclin synthase gene and essential hypertension. Hypertens Res. 2002; 25: 65-68.
- Nakayama T, Soma M, Takahashi Y, Rehemudula D, Tobe H, Sato M, et al. Polymorphism of the promoter region of prostacyclin synthase gene is not related to essential hypertension. Am J Hypertens. 2001; 14: 409-411.
- .Nakayama T, Soma M, Watanabe Y, Hasimu B, Kanmatsuse K, Kokubun S, et al. Splicing mutation of the prostacyclin synthase gene in a family associated with hypertension. Adv Exp Med Biol. 2003; 525: 165-168.
- Nakayama T, Soma M, Watanabe Y, Hasimu B, Sato M, Aoi N, et al. Splicing mutation of the prostacyclin synthase gene in a family associated with hypertension. Biochem Biophys Res Commun. 2002; 297: 1135-1139.
- Giles TD, Sander GE, Nossaman BD, Kadowitz PJ. Impaired vasodilation in the pathogenesis of hypertension: Focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens. 2012; 14: 198-205.
- Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001; 286: 954-959.
- Johnson AG, Nguyen TV, Day RO. Do nonsteroidal anti-inflammatory drugs affect blood pressure? A meta-analysis. Ann Intern Med. 1994; 121: 289-300.
- Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001; 345: 1809-1817.
- Hermida RC, Ayala DE, Mojón A, Fernández JR . Ambulatory blood pressure control with bedtime aspirin administration in subjects with prehypertension. Am J Hypertens. 2009; 22: 896-903.
- Snoep JD, Hovens MM, Pasha SM, Frolich M, Pijl H, Tamsma JT, et al. Time-dependent effects of low-dose aspirin on plasma renin activity, aldosterone, cortisol, and catecholamines. Hypertension. 2009; 54: 1136-1142.
- Trott DW, Harrison DG. The immune system in hypertension. Adv Physiol Educ. 2014; 38: 20-24.
- WHITE FN, GROLLMAN A. Autoimmune Factors Associated With Infarction of The Kidney. Nephron. 1964; 1: 93-102.
- Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol. 1982; 128: 1211-1216.
- Ryan MJ. An update on immune system activation in the pathogenesis of hypertension. Hypertension. 2013; 62: 226-230.
- Viel EC, Lemarié CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010; 298: H938-944.
- Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin ii-induced hypertension and vascular injury. Hypertension. 2011; 57: 469-476.
- Rodríguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND. Evolution of renal interstitial inflammation and NF-kappaB activation in spontaneously hypertensive rats. Am J Nephrol. 2004; 24: 587-594.
- Muller DN, Kvakan H, Luft FC. Immune-related effects in hypertension and target-organ damage. Curr Opin Nephrol Hypertens. 2011; 20: 113-117.
- Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013; 62: 126-133.
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010; 55: 500-507.
- Leavy O. T cells: Salt promotes pathogenic TH17 cells. Nat Rev Immunol. 2013; 13: 225.
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013; 496: 513-517.