
Review Article
Austin Chromatogr. 2016; 3(1): 1042.
Advanced uses of IMAC Affinity Chromatography
Allen TakYiu Lo2,3, Tan HY2,3 and Bianco PR1,2,3*
¹Department of Biochemistry, University at Buffalo, USA
²Department of Microbiology and Immunology, University at Buffalo, USA
³Center for Single Molecule Biophysics, University at Buffalo, USA
*Corresponding author:Bianco PR, Center for Single Molecule Biophysics, Department of Microbiology and Immunology, University at Buffalo, Buffalo, NY 14214, USA
Received: April 06, 2016; Accepted: May 18, 2016; Published: May 20, 2016
Abstract
Ever since the development of molecular biology, various techniques have allowed researchers to engineer proteins of interest. Isolation of that protein from crude cell lysatesoften is the rate-limiting step in protein studies. Immobilized-Metal Affinity Chromatography (IMAC) provides one of the easiest methods for protein purification. It is a robust purification method resulting in nearly homogeneous protein (free of nucleases); which is suitable for any downstream characterization and studies such as crystallization or singlemolecule experiments. In this miniview, we provide insight to improve the protein homogeneity and summarize recent advanced uses of this method to isolate protein complexes formed in vivo.
Keywords: IMAC; Affinity chromatography; MBP
Introduction
With the advances of cloning techniques, tagging proteins of interest has become a routine practice in the laboratory. There are different tags available to choose from for various purposes. Some tags are used for protein purification while others are used to track a protein in vivo. For example, fluorescent protein tags permit visualization of protein localization in the cell [1,2]. Some common tags used for purification purposes are Glutathione S-Transferees (GST) [3], Maltose-Binding Protein (MBP) [4], histidine (His) [5], and FLAG™ [6] etc. Histidine-tags (His-tag) are one of the most popular modifications used to facilitate protein purification. These tags consist of a stretch of four to ten consecutive histidine residues (with six being the most widely used [3]). They can be introduced at either the N- or C-terminus of the target protein to provide robust purification through an Immobilized Metal Affinity Column (IMAC).
This affinity column consists of a supporting matrix with an attached ligand and the immobilized metal ion. These are available from different manufacturers (Table 1). The most common supporting matrix consists of cross linked agarose beads (6%), which are large porous beads that provide high binding capacity of the resin while maintaining its stability in various pH and chemicals necessary during purification. Nitrilotriacetic Acid (NTA) is the most commonly used ligand to coordinate a metal ion on the column. It is a tetra dentate chelator that immobilizes the metal ion through four coordinate covalent bonds on the column. The remaining two coordination sites interact with the histidine side chains of the tag so that His-tagged protein can be retained on the column, which proteins lacking such a tag will flow through the column [3].
![]()
Manufacturers
Qiagen1
Sigma2
ThermoFisher Scientific3
GE Healthcare4
Clontech5
Bio-Rad6
Product Names
Ni-NTA Agarose
HIS-Select® Nickel Affinity Gel
HisPur™ Ni-NTA Superflow Agarose
HisTrap™ FF Crude
TALON® Metal Affinity Resin
Nuvia™ IMAC resins
[NaCl] and [imidazole] in washing buffer
300mM NaCl; 10mM imidazole
300 mMNaCl. 20-30 mM imidazole
300mM NaCl; 10mM imidazole
300mM NaCl; 10mM imidazole
10mM imidazole
300mM NaCl; 5mM imidazole
Ligand7
NTA
Proprietary tetradentate chelate
NTA
Proprietary chelating group
Proprietary tetradentate chelate
NTA
Resin Matrix
6% sepharose
6% beaded agarose
Crosslinked 6% agarose
Crosslinked 6% agarose
Crosslinked 6% agarose
Proprietary UNOsphere
Metal ion (alternative ion)
Ni2+
Ni2+
Ni2+(Co2+ or Cu2+)
Ni2+(Co2+ or Zn2+)
Co2+
Ni2+(Cu2+, Co2+ or Zn2+)
Suggested CVs used in the washing step
8
20
5-10
10-15
7
5
1Qiagen Quick-Start Protocol – Ni-NTA Agarose Purification of 6xHis-tagged Proteins from E. coli under Native Conditions
2Sigma-Aldrich Product Information – HIS-Select® Nickel Affinity Gel
3ThermoFisher Scientific HisPur™ Ni-NTA Super flow Agarose Instructions
4GE Healthcare HisTrap™ FF Crude, 1 mL and 5 mL Instructions
5Clontech TALON® Metal Affinity Resins User Manual
6Bio-Rad®Nuvia™ IMAC Resin Instruction Manual
7NTA: nitrilotriacetic acid
Table 1: Selected columns or resins for purification of histidine-tag proteins from various manufacturers.
HisTrap™ FF Crude columns (GE Healthcare) have been used in our laboratory for purifying His-tag proteins. This resin has a sepharose bead supporting matrix with metal chelating ligands to bind to Nickel (II) ions. In addition to the straightforward purification of histidine tagged proteins by this chromatographic resin, advanced uses have been developed for studying proteinprotein interactions and complex isolation where one partner is histagged. In this mini-review, the focus will be on the advanced uses of IMAC chromatography in our laboratory. For a more detailed review the reader is referred to [7].
Affinity Chromatography
In the Bianco laboratory, bacterial expression strains are used for over-expression of His-tagged proteins cloned into one of the available pET vectors (Novagen) [8]. In the majority of cases, the most success has been achieved using Tuner™ cells (Novagen). This strain is a specially engineered BL21 strain bearing lacY1 gene, encoding a mutant form of the membrane protein, lacpermease. This form of protein permits the inducer Isopropyl β-D-1-Thiogalactopyranoside (IPTG) to be taken up by the cells in a concentration-dependent manner. This non-able, allolactose mimic maintains constant concentration during culture growth, and binds to lac repressor thereby inactivating it, enabling T7 RNA polymerase to be expressed. The expressed polymerase then binds to the promotor upstream of the gene of interest in the pET vector, so that the gene is transcribed and the protein is expressed uniformly in all cells in the media. Different levels of expression can be achieved by varying the concentration of IPTG, typically 0.1 to 1mM [8].
Following transformation into Tuner™ cells, expression and solubility of the protein of interest are verified. Here, 5 mL cultures are grown overnight in media containing antibiotics required for plasmid selection and 0.2% glucose to repress expression to basal levels. The following day, 50 μL of each overnight is transferred to fresh media containing antibiotics only and grown for 2 hours. A sample is collected before induction as a control to compare the expression of the proteins. IPTG is then added to induce protein expression and growth is continued for an additional 3 hours. Next, two separate 1-mL aliquots of induced cells are harvested and processed to produce “Total” and a “Soluble” protein lysates (Figure 1). To produce “Total” protein lysate, one of the aliquots is subjected to centrifugation and the resulting cell pellet resuspended in water, followed by the addition of SDS to 1% (final) and lysed by boiling at 100°C for 5 minutes. This releases both soluble and insoluble proteins (T lanes in Figure 1). To produce a lysate containing only the soluble proteins, the second 1-mL aliquot of cells is subjected to centrifugation, resuspension, and lysis using either B-PER™ (Pierce) or Bugbuster® (Novagen). When equal amounts of the Total (T) and Soluble (S) lysate are compared side-by-side on a SDS-PAGE gel, solubility (or lack thereof) is readily observed (Figure 1). Culture conditions are often further optimized to maximize solubility. Thereafter, large scale growth of Escherichia coli takes place.
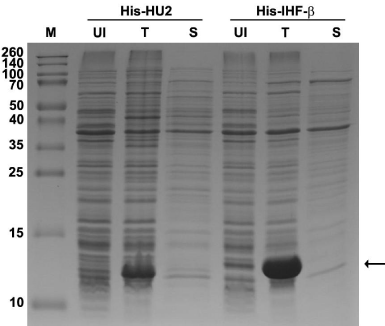
Figure 1: The His-HU2 and His-IHF-β proteins show limited solubility when
over-expressed. A Coomassie-stained, 12% SDS-PAGE gel showing the
levels of expressed proteins. Soluble protein fractions were generated by
using B-PER lysis reagent (Pierce). M: molecular weight marker; UI: uninduced
control; T: total protein; S: soluble protein fraction. The theoretical
masses of His-HU2 and His-IHF-β are 11.7 and 12.8kDa, respectively. Arrow,
expressed proteins.
The column typically used in the Bianco laboratory, HisTrap™ FF Crude (GE Healthcare), is pre packed with Ni Sepharose 6 Fast Flow resin, which consists of highly cross-linked 6% agarose beads (90 μm) with the chelating ligand immobilized on it. The Nickel (II) ion is commonly used as the immobilized metal, providing a coordinate center for binding the immobile phase of the column resin. These ions bind efficiently to histidine-tag proteins, but at the same time, proteins with clusters of histidine would bind non-specifically [9]. For resins from other manufacturers, cobalt (II) is the metal ion of choice (Table 1) [10]. The type of metal ion chelated is also dictated by the protein being purified. For example, our laboratory has had limited success purifying N-terminally His-tagged human Rad54 protein using cobalt resins whereas the nickel resin has been routinely successful (Bianco, unpublished).
The protocol of purification of His-tag proteins typically involves lysozyme/detergent lysis in the presence of benzonase nuclease and an EDTA-free protease inhibitor cocktail (Thermo Fisher Scientific or Roche) along with the serine protease inhibitor, Phenylmethylsulfonyl Fluoride (PMSF). Details can be found in the Supplementary Material. The detergents used for efficient lysis consists of a non-ionic detergent, Triton X-100 and an ionic detergent, deoxycholate. Finally, imidazole and either NaCl or KCl are added to minimize non-specific binding to the IMAC resin and to enhance protein solubility. Next, clarification of the lysate is achieved by centrifugation (40,000 x g for 30 min at 4°C). The resulting cleared cell lysate (supernatant) can be diluted using column binding buffer or loaded directly onto the nickel column.
Extensive Washing is Critical
Once loading has completed, the majority of manufacturers recommend washing the column with 5-20 Column Volumes (CVs) of binding buffer (Table 1). In contrast, in the Bianco laboratory, an extensive washing procedure involving a total of 120CVs is necessary to achieve homogeneous preparations of protein, free of contaminating nucleic acids. To achieve this, three sequential wash steps are performed. The first wash involves binding buffer for 50CVs; the second, a 40CV wash using binding buffer containing a non-ionic, non-denaturing 0.2% Nonidet P-40 (NP-40) detergent. This step is critical for the elimination of unwanted nucleic acids to less than 1% of total eluted protein, as well as contaminating proteins [11]. The third and final step is a 30CV wash using binding buffer only to remove NP-40 and to restore the UV absorbance to zero. Previously it has been found that the inclusion of a wash step with a buffer containing a low concentration of EDTA (0.5mM) can also eliminate non-specific protein binding to the resin [12]. A higher initial imidazole concentration (70mM) could also be used to minimize non-specific binding provided that the protein of interest binds to the column at this concentration [13].
Elution of Protein Depends on Both the Salt and Imidazole Concentrations
Once washing has concluded, elution commences. Although this can be achieved by washing the column with buffer containing 500mM imidazole (step elution), significantly improved results can be achieved using a linear gradient from 30 to 500mM imidazole. For the majority of proteins and protein complexes, a linear, 20CV gradient of is employed. Generally, proteins containing a single hexa-histidine tag elute at lower concentrations of imidazole (up to 200mM) whereas proteins such as the homo-tetrameric E. coli Single- Stranded DNA Binding Protein (SSB) require 350-400mM imidazole [11]. Furthermore, the His-tagged T4polynucleotide kinase requires imidazole concentrations as high as 1.3 M for elution [14].
In addition to changing concentrations of imidazole to affect protein elution, both the type and concentration of monovalent cation in the elution buffer can also be taken advantage of. Some manufacturers recommend buffers with either 500mM NaCl or 600mM KCl while others recommend lower salt concentration (Table 1). When switching between Na+ and K+ ions, it is important to also alter the buffer from sodium to potassium phosphate, so that the conductivity of the buffer is solely contributed from one type of cation. In addition to the type of monovalent cation present, the [cation] can also have a marked effect on the amount and quantity of protein eluted as well a son complex stability (described later). Finally, different concentrations of monovalent cations can alter the concentration of imidazole required to elute bound proteins [11]. For example, when the elution profiles of the His-SSB/RecG complex in 150 or 600mM KCl containing buffers are compared, it is clear that the protein peaks at different points in the imidazole gradient (Figure 2a). In 150mM KCl buffer, the peak is centered at 471mM imidazole, whereas in 600mM KCl, the complex elutes earlier at 300mM imidazole (Figures 2b and c).
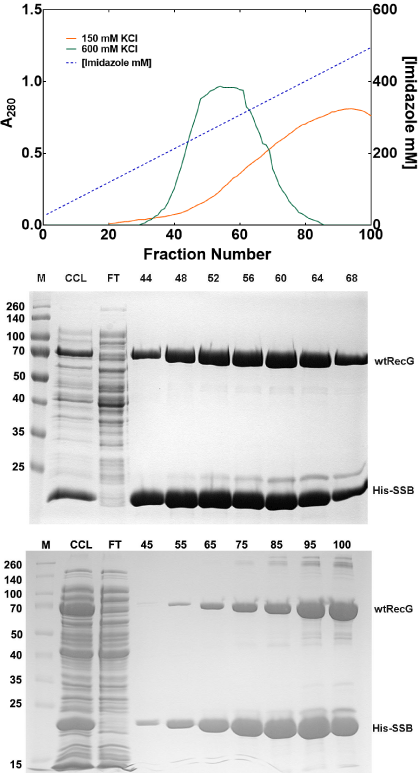
Figure 2: Monovalent cation concentrations affect protein elution conditions.
(A) The elution position of the His-SSB/wtRecG complex is affected by
monovalent cation concentration. Two separate purifications were done
on separate days. (B) and (C), Coomassie-stained 12% SDS-PAGE gels
of the purifications yielding the elution profiles in panel A. (B), elution in
buffer containing 600 mMKCl. (C) Elution in buffer containing 150 mMKCl.
M: molecular weight marker; CCL, cleared cell lysate; FT, flow-through.
Numbers above the gels correspond to the fraction numbers from the elution
profiles shown in panel A. The graph and corresponding gels appeared in [11]
and are used by permission of John Wiley and Sons.
Advanced techniques of nickel column chromatography
In addition to the purification of His-tag proteins, IMAC can also be taken advantage of in isolating chimeric proteins with different ratios of auto fluorescent protein incorporation; to purify hetero-oligomeric protein complexes, or to study protein-protein interactions. Details of each are discussed below:
Isolation of chimeric, multi-subunit proteins
Fluorescent labelling of proteins, by fusing them to GFP for example, allows the observation of protein localization in vivo and can provide additional insight into function. In the case of multimeric protein complexes, the incorporation of multiple GFPs into the complex may compromise function. This is particularly true for RuvA and the essential, Single Stranded DNA Binding protein (SSB). Both proteins exist as homotetramers in solution [15,16). RuvAB is responsible for branch migration of Holliday junctions, the central intermediate recombination [17,18]. RuvA binds to the junctions and loads RuvB hexamers onto opposing arms [19]. SSB is involved in all aspects of DNA metabolism where it binds to single-stranded DNA intermediates and recruits repair and replication proteins to process the intermediates. Binding to target proteins is mediated via the essential C-terminus of SSB [20].
Our laboratory recently developed a dual plasmid expression system to produce chimeras of the SSB and RuvA proteins, as the homotetrameric tagged versions were inactive [21,22]. Consequently, in each case, one plasmid encodes a histidine-tagged subunit while the other encodes a subunit fused in-frame to GFP (or any other auto fluorescent protein). The dual plasmid expression system allows us to produce mixed species of tetramer proteins in the same cell. The introduction of a histidine tag provides robust purification while introducing an auto fluorescent protein allows us to observe localization of the tetrameric protein. Both of these features can coexist in a single chimeric, tetramer.
By expressing proteins from the dual plasmids in a single cell, a heterogeneous population of chimeras is produced. These contain zero, one, two, three or four histidine tags and correspondingly, four, three, two, one or zero GFP-fusion subunits. The varying numbers of histidine tags are then taken advantage of during elution. First, a homo-tetrameric GFP-fusion which contains no histidine tag does not bind to the column and is found in the flow through. Once washing has concluded, and the elution gradient applied, chimeras with a single His-tag elute first with increasing concentrations of imidazole from the gradient resulting in the elution of chimeras with more tags (Figure 3a). When the resulting species are analyzed by SDS-PAGE, the composition of the purified chimeras is evident (Figure 3b). Further analysis of chimera composition by gel filtration on a Superpose 6 column confirmed the gel results. As the number of GFP-fusion subunits increased, the chimeras eluted earlier relative to wild type, consistent with the increase in associated molecular weight (Figure 3c). Satisfyingly, the activity of chimeric species containing one or two fusion subunits was identical to wild type, while those with three GFPs had reduced activity [21,22]. Similar success was achieved with RuvA [22].
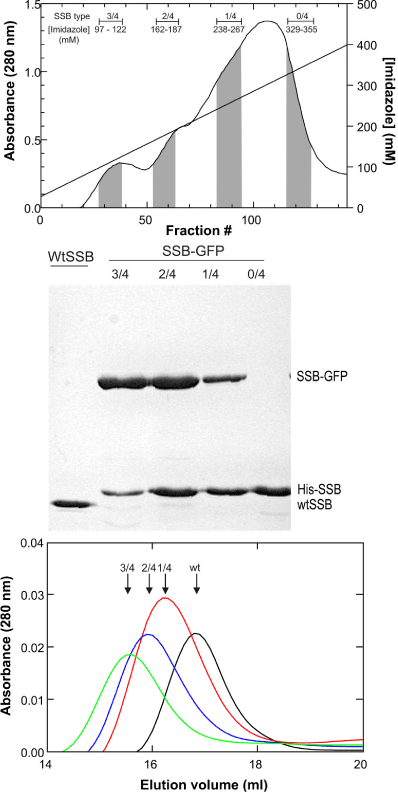
Figure 3: Careful gradients facilitate isolation of distinct chimeric species.
(A), Elution profile of SSB-GFP/His6-SSB chimeras. In this experiment, a
20mL Ni-sepharose high performance column (GE Healthcare) was used.
Due to the different number of histidine tags present, chimeras eluted
at different times during the gradient. The designation 3/4 corresponds to
3SSB-GFP/1His6-SSB; 2/4 corresponds to 2SSB-GFP/2His6-SSB; 1/4
corresponds to 1SSB-GFP/3His6-SSB; 0/4 corresponds to 4His6-SSB only.
(B), A Coomassie-stained, 12% SDS-PAGE gel of the final pool of each
chimera. (C), Gel filtration elution profiles of each chimera. In this experiment,
a 24mLSuperose 6 column was used. These data appeared in [21] and are
used by permission of John Wiley and Sons.
Isolation of protein interaction partners
In a similar fashion to the purification of chimeras, the binding of one protein to another in vivo can also be characterized [23], provided the complex remains intact throughout lysis and subsequent chromatographic steps [7]. To characterize binding, the dual plasmid system is again employed. However, in this case the first plasmid encodes His-SSB while the second encodes a target protein, in this case wild type RecG helicase. The tagging scheme can also be reversed in separate experiments [11,21]. Results show that His-SSB and RecG co-elute during the imidazole gradient (Figure 2). Intriguingly, by altering the concentration of KCl in the elution buffer, insight into the nature of the interactions being used to form the complex can be obtained. As more of the SSB-RecG complex elutes in buffer containing 150mM KCl than in 600 mM KCl , this indicates that a significant component of the interaction is electrostatic in nature. The reverse would be true if the interaction were dominated predominately by hydrophobic effects.
Isolation of hetero-multimeric complexes
Finally, nickel column chromatography can also be combined with additional tagging and chromatography techniques for isolating the hetero-multimeric complexes. Several groups have also adapted this method to isolate protein complexes with only one of the proteins in the complex being His-tagged [23-25]. In our laboratory, we focused on a protein complex required for stalled DNA replication fork rescue. This complex consists of a single SSB tetramer simultaneously bound to the repair helicases PriA and RecG.
To determine if this complex exists in vivo, a triple plasmid
system was employed. Here, SSB was N-terminally, Profinity tagged
(Pro-SSB), RecG was His-tagged at the C-terminus (RecG-His),
and PriA was C-terminally biotin tagged (PriA-Bio). The resulting
clarified lysate was subjected to IMAC, Profinity (Bio-Rad), and
monomeric avidin column chromatography sequentially [11,26].
Profinity chromatography takes advantage of the fusion of a small
N-terminal affinity tag (the Profinity tag; 8 kDa) to the protein of
interest that is recognized by a resin with mutant subtilisin proteinase
ligand. These bind with very high affinity (dissociation constant less
than 100pM), with the release of the protein of interest achieved by
washing with 100mM potassium fluoride. This facilitates cleavage of
the tag by subtilsin proteinase (ProfinityeXact™ Protein Purification
System - Instruction Manual, Bio-Rad). The monomeric avidin
column utilizes affinity between biotin and avidin. In the native
avidin tetramer, biotin binding occurs with high affinity (dissociation constant = 10
The experimental scheme is shown in (Figure 4). First, when cells are lysed, four possible complexes are released (RecG-His/Pro-SSB, PriA-Bio/Pro-SSB, PriA-Bio/RecG-His, and RecG-His/Pro-SSB/ PriA-Bio). As only RecG is histidine-tagged, any protein(s) bound to it will co-elute from the nickel column. When the eluted mixture of RecG-His/Pro-SSB, PriA-Bio/RecG-His, and RecG-His/Pro-SSB/ PriA-Bio is applied to the ProfinityeXact™ column, only proteins in complex with SSB bind to the resin. Once the profinity tag is cleaved, the RecG-His/SSB and RecG-His/SSB/PriA-Bio complexes are eluted (Right panel of Figure 4). The identification of PriA-Bio by biotin blots in the final elution, confirmed the presence of the three protein complex (RecG-His/Pro-SSB/PriA-Bio). It is worth noting here that for this experiment to be successful, the concentration of NaCl in the lysis and column buffers had to be reduced to 100mM to maintain complex stability (Figure 2 and [7]).
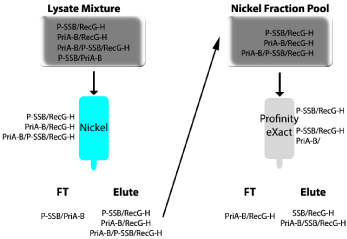
Figure 4: Purification of a triple-tagged, three-protein complex.
A schematic showing the purification scheme and eluted complexes is shown.
RecG was C-terminal His-tagged (RecG-H), SSB was N-terminal, Profinitytagged
(Pro-SSB), and PriA was biotinylated at the C-terminus (PriA-Bio).
The cleared cell lysate produced from cells expressing all three plasmids
was applied to the Nickel column. Complexes that could bind to the resin are
indicated in cyan on the left or right of each column. The eluted complexes
were applied directly to the ProfinityeXact™ column (grey; right panel of the
Figure) and eluted as described.
Summary
Immobilized metal affinity column chromatography is a robust technique. One-column purification can produce homogeneous preparations of histidine-tagged proteins free from contaminating nucleic acids and other proteins. This high quality protein facilitates achieving high quality data from various studies including singlemolecule. This chromatographic technique can also be used in novel ways to isolate differentially-tagged multi-subunit complexes and can be combined with other tagging and chromatographic techniques. The isolated protein complex gives us precious information on protein-protein interactions in vivo.
References
- Bianco PR, Stanenas AJ, Liu J, Cohan CS. Fluorescent single-stranded DNA-binding proteins enable in vitro and in vivo Studies, in Single-Stranded DNA Binding Proteins. Keck JL, Editor. 2012; 922: 235-244.
- Remington SJ. Green fluorescent protein: a perspective. Protein Sci. 2011; 20: 1509-1519.
- Schafer F, Seip N, Maertens B, Block H, Kubicek J. Purification of GSTTagged Proteins. Methods Enzymol. 2015; 559: 127-139.
- Duong-Ly KC, Gabelli SB. Chapter Two - Affinity Purification of a Recombinant Protein Expressed as a Fusion with the Maltose-Binding Protein (MBP) Tag, in Methods Enzymol. Jon RL, Editor. 2015; 599: 17-26.
- Spriestersbach A, Kubicek J, Schafer F, Block H, Maertens B. Purification of His-Tagged Proteins. Methods Enzymol. 2015; 559: 1-15.
- Gerace E, Moazed D. Affinity Pull-Down of Proteins Using Anti-FLAG M2 Agarose Beads. Methods Enzymol. 2015; 559: 99-110.
- Block H, Maertens B, Spriestersbach A, Brinker N, Kubicek J, Fabis R, et al. Reprint of: Immobilized-Metal Affinity Chromatography (IMAC): A Review. Protein Expr Purif. 2011.
- Casali N. Escherichia coli host strains, in E. coli Plasmid Vectors. Casali N, Preston A, Editors. 2003, Humana Press. 27-48.
- Ristic D, Modesti M, van der Heijden T, van Noort J, Dekker C, Kanaar R, et al. Human Rad51 filaments on double- and single-stranded DNA: correlating regular and irregular forms with recombination function. Nucleic Acids Res. 2005; 33: 3292-3302.
- Chaga G, Hopp J, Nelson P. Immobilized metal ion affinity chromatography on Co2+-carboxymethylaspartate-agarose Superflow, as demonstrated by one-step purification of lactate dehydrogenase from chicken breast muscle. Biotechnol Appl Biochem. 1999; 29: 19-24.
- Yu C, Tan HY, Choi M, Stanenas AJ. SSB binds to the RecG and PriA helicases in vivo in the absence of DNA. Genes Cells. 2016; 21: 163-184.
- Westra DF, Welling GW, Koedijk DG, Scheffer AJ, The TH, Welling-Wester S. Immobilized metal-ion affinity chromatography purification of histidinetagged recombinant proteins: a wash step with a low concentration of EDTA. J Chromatogr B Biomed Sci Appl. 2001; 760: 129-136.
- Ichikawa S, Takai T, Yashiki T, Takahashi S, Okumura K, Ogawa H, et al. Lipopolysaccharide binding of the mite allergen Der f 2. Genes Cells. 2009; 14: 1055-1065.
- Wang LK, Shuman S. Domain structure and mutational analysis of T4 polynucleotide kinase. J Biol Chem. 2001; 276: 26868-26874.
- Krauss G, Sindermann H, Schomburg U, Maass G. Escherichia coli singlestrand deoxyribonucleic acid binding protein: stability, specificity, and kinetics of complexes with oligonucleotides and deoxyribonucleic acid. Biochemistry. 1981; 20: 5346-5352.
- Tsaneva IR, Illing G, Lloyd RG, West SC. Purification and properties of the RuvA and RuvB proteins of Escherichia coli. Mol Gen Genet. 1992; 235: 1-10.
- Bianco PR. DNA Helicases. eLS. John Wiley & Sons, Ltd: Chichester. 2012.
- Bianco PR. DNA Helicases. EcoSal Plus. 2010; 4.
- Parsons CA, Stasiak A, West SC. The E.coli RuvAB proteins branch migrate Holliday junctions through heterologous DNA sequences in a reaction facilitated by SSB. EMBO J. 1995; 14: 5736-5744.
- Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol. 2008; 43: 289-318.
- Liu J, Choi M, Stanenas AG, Byrd AK, Raney KD, Cohan C, et al. Novel, fluorescent, SSB protein chimeras with broad utility. Protein Sci. 2011; 20: 1005-1020.
- Tan HY, Wahab SA, Seet JX, Bianco PR. Construction, purification and characterization of novel, fluorescent RuvA chimeras. Austin Chromatogr. 2015; 2: 1027.
- Lougheed KE, Bennett MH, Williams HD. An in vivo cross linking system for identifying mycobacterial protein-protein interactions. J Microbiol Methods. 2014; 105: 67-71.
- Li L, Patterson DP, Fox CC, Lin B, Coschigano PW, Marsh EN. Subunit structure of benzylsuccinate synthase. Biochemistry. 2009; 48: 1284-1292.
- Diaz-Perez C, Rodriguez-Zavala JS, Diaz-Perez AL, Campos-García J. Co-expression of a and β subunits of the 3-methylcrotonyl-coenzyme A carboxylase from Pseudomonas aeruginosa. World J Microbiol Biotechnol. 2012; 28: 1185-1191.
- Martini VP, Glogauer A, Müller-Santos M, Iulek J, de Souza EM. First coexpression of a lipase and its specific foldase obtained by metagenomics. Microb Cell Fact. 2014; 13: 171.
- Morag E, Bayer EA, Wilchek M. Reversibility of biotin-binding by selective modification of tyrosine in avidin. Biochem J. 1996; 316: 193-199.