
Research Article
Austin J Clin Immunol. 2014;1(2): 1007.
Vaccination with the Prostate Cancer Over-Expressed Tumor Self-Protein TPD52 Elicits Protective Tumor Immunity and a Potentially Unique Subset of CD8+ T Cells
Jennifer D Bright1, Joel F Aldrich1, Jennifer A Byrne2 and Robert K. Bright1,*
1Department of Immunology and Molecular Microbiology, Texas Tech University Health Sciences Center
2Children’s Cancer Research Unit, University of Sydney Discipline of Pediatrics and Child Health
*Corresponding author: Robert K. Bright, Department of Immunology and Molecular Microbiology, Texas Tech University Health Sciences Center, 3601 4th Street, MS 6591, Lubbock, TX 79430
Received: January 16, 2014; Accepted: February 10, 2014; Published: February 17, 2014
Abstract
Tumor protein D52 (D52) is expressed at low levels in normal cells, but over-expressed in prostate carcinomas and numerous other malignancies. Murine D52 (mD52) parallels the expression pattern of the human orthologue (hD52) and shares ~ 86% amino acid identity. Over-expression of mD52 in nontransformed murine fibroblasts induces anchorage independent growth and spontaneous metastasis. The TRAMP model was employed to study DNA-based D52 vaccines against prostate cancer. Immunizations consisted of mD52-DNA, hD52-DNA or a combination of both, followed by challenge with mD52 positive, TRAMP-C1 tumor cells. Greater protection (70%) was observed 10 months post challenge in mice immunized with hD52 DNA. Survivors of the initial tumor challenge rejected a second tumor challenge with mD52 positive, autochthonous TRAMP-C2 tumor cells given in the opposite flank more than four months after the first challenge. Analysis of the T cell function from survivors indicated that a Th1-type cellular immune response was involved in tumor rejection. A potentially unique subset of CD8+ IL-10+ T cells was also elicited and may play a role in inhibiting vaccine induced tumor immunity, suggesting that a deeper mechanistic understanding of these T cells in D52 vaccine-induced immunity may be important for developing a more potent cancer vaccine.
Keywords: Prostate; Vaccine; hD52; mD52; TPD52; Murine; TRAMP; CD8+ regulatory T cells.
Abbreviations
TPD52: Tumor protein D52; mD52: murine TPD52; hD52: human TPD52; TAA: tumor associated antigen; T regulatory cell, Treg
Introduction
A 2009 National Cancer Institute sponsored project to prioritize cancer vaccine target antigens for translational-research revealed that over expressed tumor self-proteins represent the largest number of untested antigens for vaccine development [1]. While it is arguable that antigens that are only found in tumors and not normal cells should be “ideal” targets for vaccination, most cancer antigens that have been isolated from tumor cells to date are self-proteins, specifically they are expressed at low levels in normal cells and over expressed in tumor cells, a characteristic that facilitated their discovery [2]. Until now only Her-2/neu could be classified as an over expressed tumor self-antigen that demonstrates a role in oncogenicity. This property has been proposed to be a desired and important characteristic for the next generation of cancer vaccine target antigens [1]. We recently described a novel over expressed tumor self-antigen, tumor protein D52 (D52). D52 represents a shared tumor antigen with a wide range of cancer associations to include but not limited to breast, prostate and ovarian cancers [3], and like Her-2/neu, D52 exhibits oncogenic properties [4,5].
Tumor protein D52 is a naturally expressed intracellular protein present at low but detectable levels in healthy cells and tissues where its normal function has yet to be defined. Increased expression of D52 has been demonstrated in association with prostate cancer as well as numerous other human malignancies [6-22]. The murine orthologue of D52 (mD52) is ~ 86% identical to human D52 (hD52) at the amino acid level [23]. Previous work from our laboratory demonstrated that over expression of mD52 in normal murine fibroblast cells induced anchorage independent growth in vitro and spontaneous lung metastasis in vivo [5]. We also demonstrated that reduction of hD52 expression via RNAi resulted in increased apoptosis in human breast cancer cells and hD52 over-expression correlated with decreased survival in human breast cancer patients [4]. Interestingly, shRNA reduction of mD52 expression abrogates spontaneous metastasis associated with murine 3T3.mD52 sarcoma cells [unpublished observation]. Thus, D52 is actively involved in transformation, leading to increased cell proliferation and metastasis. Involvement in oncogenesis suggests that these antigens may be critical for tumor survival, making the over expressed tumor self-protein D52 an excellent candidate for a cancer vaccine target.
Herein, we tested the hypothesis that the xenogeneic human orthologue of D52 (hD52) when administered i. m. as a simple DNAbased vaccine would elicit an anti-tumor immune response that is more potent than that elicited by the mD52 as assessed by protection from challenge with autochthonous TRAMP-C tumor cells which naturally contain elevated levels of mD52 protein [24].
Materials and Methods
Mice and tumor cell lines
Male 6- to 8-week old C57BL/6 mice were purchased from Jackson Labs (Bar Harbor, ME). All animals were cared for and treated according to Institutional Animal Care and Use Committee (IACUC) guidelines at Texas Tech University Health Sciences Center (Lubbock, TX). All experiments were conducted with IACUC approval. The tumorigenic, autochthonous C57BL/6 cell lines TRAMP-C1 and TRAMP-C2 [25] were used for tumor challenge and as targets for immunoassays. The tumorigenic SV40-transformed Balb/c murine kidney cell line designated mKSA was used as an mD52-positive MHC mis-matched control target for immunoassays [24,26,27].
Purification and validation of plasmid DNA used for immunization
Luria- Bertani (LB) broth supplemented with ampicillin (100ug/ ml) was inoculated with a starter culture of JM109 bacterial cells transformed with pcDNA, mD52pCDNA or hD52pcDNA plasmid and grown overnight with shaking (300 rpm) at 37oC. Bacterial cells were lysed, and plasmid DNA purified using Qiagen’s Endo Free Plasmid Purification kit (Valencia, CA) according to the manufacturer’s instructions. DNA concentrations were calculated using a bio-photometer (Eppendorf, Westbury, NY) [26]. Restriction enzyme digests were performed on all plasmids to confirm the presence of mD52 or hD52 cDNA inserts. Endpoint PCR was performed using primers specific for mD52 or hD52 for 30 cycles to confirm presence of the respective cDNA insert. Primer sequences: mD52cds-F-5’- TGC TGA AGA CAG AGC CGG, mD52cds-R-5’- ACG TCT TGC CAC CCT TTG, hD52-F-5’- GAT CTC GGG CTG GAG ACA TGG, hD52-R-5’- AAT TCG TGG GTA GCA GAA CAA AGG. Annealing temperatures used were 62oC and 60oC for mD52cds and hD52 primers, respectively. Primers for GAPDH were used as an internal control reference in PCR experiments [5]. To confirm mD52 and hD52 protein expression 3T3 cells were transfected with the vaccine plasmids containing cDNAs for mD52 or hD52, and whole cell protein lysates were prepared using methods previously described [4,28]. Protein expression was detected by Western analysis using an anti-TPD52 polyclonal antibody (generated by immunizing rabbits with N-terminal, carrier conjugated peptide GCAYKKTSETLSQAGQKAS; italics represents a region of TPD52 protein that is conserved between human and mouse) (Bio Synthesis, Inc, Lewisville, TX) [5].
Immunization and tumor cell challenge
Individual mice were immunized with 50 micrograms of D52- DNA administered i.m. in saline every 10 days for a total of 4 injections. Empty vector DNA (pCDNA 3.1 vector minus mD52 cDNA) served as a control immunization. Two weeks following the final immunization, mice in all groups were challenged with a tumorigenic dose (5x106) of autochthonous TRAMP-C1 tumor cells [25]. Mice that survived the primary challenge were re-challenged in the opposite flank with 1x106 TRAMP-C2 cells [25] approximately 150 days after the initial challenge. For some experiments, the TRAMP-C2 challenge dose was 5 X 105 cells, which was determined empirically to be 100% tumorigenic. Tumor size was determined by taking perpendicular measurements with calipers every 2 to 3 days and tumor volume (mm3) was calculated using the following formula: (a x b2) / 2, where b was the smaller of the two measurements [26,27].
To assess a role for CD4+ CD25+ or CD8+ CD122+ regulatory T cells in response to mD52 DNA vaccination, mice were injected i.p. with 300 μg of anti-CD25 mAb (PC-61.5.3), or anti-CD122 mAb (TM-beta 1) ), or both in 200 μl PBS on day 0, and again on day 28 at the time of the first and third mD52-DNA immunizations. At the time of tumor challenge (day 58), mice were injected i.p. with 600 μg of anti-CD25 mAb, or anti-CD122 mAb, or both in 200 μl PBS. For control (mock) depletions, mice were injected i.p. with isotype matched IgG on day 0, day 28 and at the time of tumor challenge with 300 μg, 300 μg and 600 μg, respectively.
T cell culture and ELISAs for cytokine production
T cells from immunized mice were stimulated in vitro by culturing Lympholyte-M® gradient separated spleen-derived lymphocytes with irradiated tumor cells (the same tumor cell line used for the in vivo challenge) in the presence of IL-2 (10 ng/ml), IL-7 (5 ng/ml), and IL-12 (5 ng/ml) at 37°C for 5-7 days. Culture supernatants used for cytokine analyses were harvested from 24 hr cultures of T cells (1x106 cells / ml in 200 μl of medium in 96 well plates) in medium alone, compared to T cells cultured with various tumor cell targets (1:1 ratio). Experimental targets were the TRAMP-C1 tumor cells (H-2b+, mD52+) and TRAMP-C2 tumor cells (H-2b++, mD52+). mKSA (H-2d+, mD52+) tumor cells, served as a control MHC mismatched, antigen positive target. Yac-1 cells served as an MHC-I negative control. To confirm MHC-I restricted tumor recognition, blocking assays were performed by incubating tumor cells with anti-H-2b or anti-H-2d (negative control) mAb, prior to incubation with T cells. Assessment of cytokine secretion by tumor-specific T cell cultures was accomplished by applying culture supernatants to commercially available sandwich ELISA’s for IFN-γ, IL-10, IL-4, and IL-17 detection (R&D Systems, Minneapolis,MN) as per the manufacturer’s instructions. Assays were analysed using a Victor3™ plate reader (Perkin Elmer, Boston, MA). We performed all assays with the manufacturer’s provided internal controls, from which standard curves were generated in order to determine concentration of cytokines produced in experimental sets for ELISA detection of IFN-γ, IL-10, IL-4, and IL-17 [26, 27].
Analysis of cytotoxic T lymphocyte (CTL)-mediated tumor cell lysis
T cells from spleens of immunized mice that survived tumor challenge were isolated and subjected to standard cytotoxic T lymphocyte (CTL)-mediated tumor cell lysis analysis. CTLs were generated by culturing spleen cells in the presence of irradiated tumor cells (using the same tumor cell line as was used for the in vivo challenge) in the presence of IL-2 (10 ng/ml), IL-7 (5 ng/ml), and IL- 12 (5 ng/ml) at 37°C for 5-7 days. Specificity was evaluated by mixing various numbers of CTLs with a constant number of target cells (5 x 103 cells per well) in 96 well round bottom plates. Specific lysis was determined using either a Europium time-resolved fluorescencebased method or LDH-release method, and measured using a Victor3™ plate reader (Perkin Elmer, Boston, MA) using previously described methods [27, 28, 29].
Flow cytometry
Lymphocytes from spleens cultured in vitro as described above were stained with monoclonal antibodies specific for CD3, CD4, CD19 and CD8. MHC class-I expression was assessed on tumor cell lines. Antibodies were purchased from BD-Bioscience (San Jose, CA). For determination of CD25+ Treg cell depletion, peripheral blood lymphocytes (PBLs) were collected 7 days following the 4th immunization via tail vein bleed, and lymphocytes were isolated using Lympholyte-M® density separation medium (Cedarlane Labs, Burlington, NC). Lymphocytes from animals in the same experimental group were pooled (n=10 per pooled sample) and stained with 1 μg each of anti-CD4-FITC and anti-CD25-PE or anti-CD122-PE mAbs per 1 x 106 cells. Antibodies were purchased from BD-Bioscience (San Jose, CA). Cells were fixed in 1% paraformaldehyde at 4oC for 1 hr and then analyzed by flow cytometry using a BD LSRII flow cytometer [26,27].
Real-time RT-PCR
Total RNA was extracted from pure T cell cultures that were generated by 7 day in vitro stimulation of lymphocytes from D52- DNA immunized mice with CD3, CD28 activation beads according to the manufacturer’s instructions (Life Tech, InVitrogen). For real-time RT-PCR, cDNA was generated using 1 μg of total RNA and oligo-dT primer and PCR reactions were performed using our previously published methods [5]. Primer sets for the targets depicted in figure 6b were purchased from Realtimeprimers.com (https://www.realtimeprimers.com/real-time-pcr-primer-sets-mouse-pcr-primersets.html). Reactions were performed using the Applied Bio systems Step One Plus Sequence Detection System and ABI SYBR green PCR core reagents kit, according to the manufacturer’s instructions (Applied Bio systems, Foster City, CA). PCR conditions for 40 cycles were: 60oC for 2 min, 95oC for 10 min, 95oC for 15 s, for all primers. Additional controls involved no template and no RT enzyme and were included in all real-time PCR reactions.
Statistical analysis
ELISA data were analyzed using one-way ANOVA with Bonferoni multiple comparison post test. A p value of less than 0.05 was determined to be statistically significant (Graph Pad Prism 5.0). Specific lysis data for CTL assays were analyzed using one-way ANOVA with Tukey-Kramer post test. A p value less than 0.05 was determined to be significant (Graph Pad Prism 5.0). Tumor challenge data were analyzed with a t-test to determine whether significant differences existed between mean tumor volume for D52-DNA immunized and control immunized mice [27].
Results
Intramuscular D52-DNA vaccination induces protective tumor immunity
Previously we reported on DNA-based vaccination against D52 using the TRAMP model of prostate cancer for which mD52-DNA was injected s.c. admixed with murine GM-CSF protein [26]. Though partial protection was observed following autochthonous TRAMP-C tumor challenge (40 % protected > 90 days), we postulated that vaccine approaches with xenogeneic hD52 might increase tumor rejection efficacy. To simplify the approach, we immunized mice i.m. with either hD52- DNA, mD52-DNA, hD52-DNA followed by mD52-DNA or mD52-DNA followed by hD52-DNA, and then challenged mice with a tumorigenic dose of TRAMP-C1 cells (Figure 1A). All four DNA vaccine approaches protected mice from tumor challenge. The majority of mice (70%) that received four injections of hD52-DNA remained free from TRAMP-C1 tumor growth for nearly eight months (Figure 1B), compared to 50% of mice that received four injections of mD52 prior to tumor challenge (Figure 1C). Similar tumor protection results were obtained for the primeboost approach, where mice received either two injections of hD52- DNA followed by two injections of mD52-DNA (60%) (Figure 1D) or two injections of mD52-DNA followed by two injections of hD52- DNA (70%) (Figure 1E). Interestingly, the average onset of tumor growth was delayed by nearly 21 days in mice that were immunized with hD52-DNA compared to the other vaccine groups (Figure 1CE). Overall the protective tumor immunity induced by i.m. D52-DNA vaccination was considerable and did not vary greatly whether the vaccine consisted of hD52-DNA, mD52-DNA or a combination of both. Control vaccines with empty vector DNA did not protect against tumor challenge (not shown) [26].
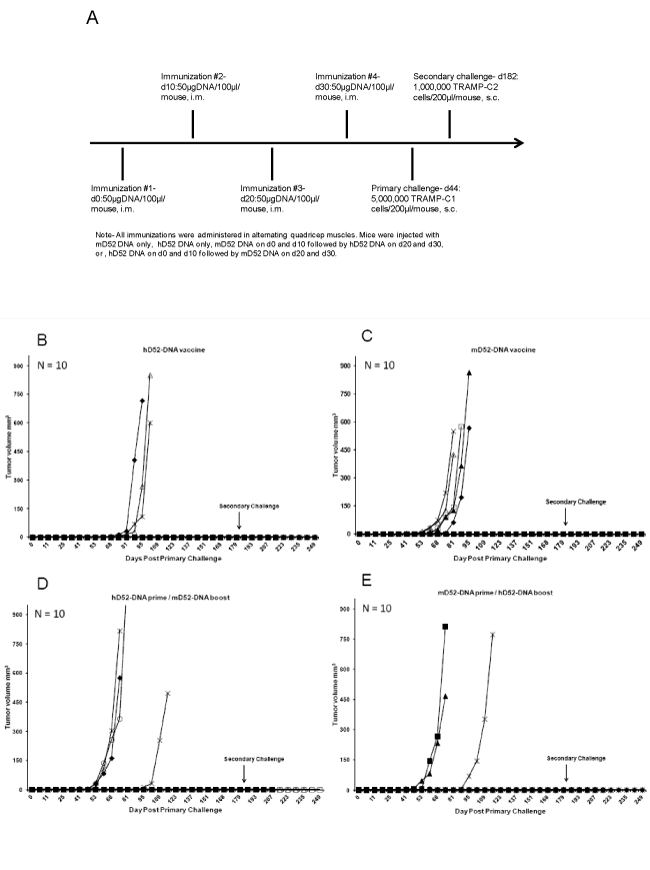
Figure 1: Tumor protection in mice immunized with D52 DNA. A) Groups of male C57BL/6 mice were immunized with 50 micrograms of D52-DNA administered i.m. in saline every 10 days for a total of 4 injections. Two weeks after the 4th injection the mice were challenged s.c. with autochthonous 5 x 106 TRAMP-C1 tumor cells. B) Mice were immunized with human D52 (hD52)-DNA and challenged with TRAMP-C1 tumor cells. C) Mice were immunized with murine D52 (mD52)-DNA then challenged with TRAMP-C1 tumor cells. D) Mice were immunized with hD52-DNA twice followed by mD52-DNA twice then challenged with TRAMP-C1 tumor cells. E) Mice were immunized with mD52-DNA twice followed by hD52-DNA twice then challenged with TRAMP-C1 tumor cells. Tumor size was determined by taking perpendicular measurements with calipers every 2 to 3 days and tumor volume (mm3) was calculated using the following formula: (a x b2) / 2, where b was the smaller of the two measurements.
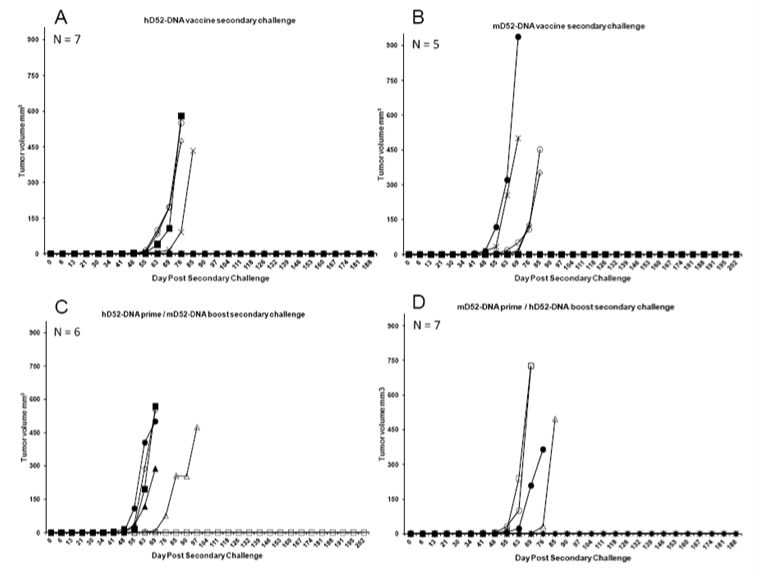
Figure 2: Immunization with D52 DNA induces protection against secondary tumor challenge. Four to five months after the rejection of a primary challenge with TRAMP-C1 tumor cells, mice were challenged s.c. with 1 x 106 autochthonous TRAMP-C2 tumor cells in the opposite flank and tumor growth was monitored. A) Mice originally immunized with human D52 (hD52)-DNA and rejected a primary tumor challenge received a secondary s.c. challenge with TRAMP-C2 tumor cells on day 182 approximately 138 days after primary challenge. B) Mice originally immunized with murine D52 (mD52)-DNA and rejected a primary tumor challenge received a secondary s.c. challenge with TRAMP-C2 tumor cells on day 182 approximately 138 days after primary challenge. C) Mice originally immunized with hD52-DNA twice followed by mD52-DNA twice and rejected a primary tumor challenge received a secondary s.c. challenge with TRAMP-C2 tumor cells on day 182 approximately 138 days after primary challenge. D) Mice originally immunized with mD52-DNA twice followed by hD52-DNA twice and rejected a primary tumor challenge received a secondary s.c. challenge with TRAMP-C2 tumor cells on day 182 approximately 138 days after primary challenge. Tumor size was determined by taking perpendicular measurements with calipers every 2 to 3 days and tumor volume (mm3) was calculated using the following formula: (a x b2) / 2, where b was the smaller of the two measurements.
To test the durability of the four different D52-DNA vaccine strategies we challenged mice that rejected TRAMP-C1 tumors with TRAMP-C2 tumor cells in the opposite flank approximately 140 days after the primary challenge, and 180 days after the final D52-DNA injection. TRAMP-C2 cells grow more aggressively than TRAMP-C1 in vivo, and so were chosen as a more stringent test of vaccine durability against a recurrent tumor. Mice that received either hD52-DNA only or mD52-DNA followed by hD52-DNA prior to primary TRAMP-C1 tumor challenge were better protected (>40%) from secondary TRAMP-C2 tumor challenge (Figures 2A and 2D), compared to mice that received mD52-DNA only (20% protected) or hD52-DNA followed by mD52-DNA (<20% protected) prior to primary tumor challenge (Figures 2B and C). Nonetheless, these data demonstrate that plasmid DNA-based vaccines targeting tumor protein D52 when delivered i.m. protected mice from s.c. autochthonous TRAMP-C1 tumor challenge. More importantly, the D52-DNA vaccine-induced immunity was durable, capable of providing protection from recurrent tumors as demonstrated by rejection of a secondary challenge with more aggressive TRAMP-C2 tumor cells administered s.c. in the opposite flank several months after the initial tumor challenge. DNA vaccines comprised of the human orthologue of D52 (hD52) appeared to be slightly more potent than mouse D52 (mD52) although both vaccine approaches demonstrated efficacy. It is important to note that both TRAMP-C1 and TRAMP-C2 tumor cells naturally over express mD52 protein without experimental manipulation [24], and were not modified to express hD52.
hD52-DNA vaccination elicits CD8+ CTLs and IL-10 producing CD8+ T cells
In other studies, we reported that mD52-DNA administered s.c. [26], mD52 protein administered i.m. [24], and mD52 protein administered s.c. [27] elicited Th1-type T cell immunity characterized by the production of IFN-γ and CTL killing of the challenging tumor cells. These studies were focused on mD52 vaccines only. Since we demonstrated efficacy of hD52-DNA vaccination here for the first time, and knowing that D52 is an intracellular protein, we were interested in interrogating the MHC class-I-restricted T cells response involved in tumor protection following hD52-DNA vaccination. Splenocytes were harvested from mice that were immunized with hD52-DNA and survived both a primary and a secondary tumor challenge. The effector cells generated by 5-7 day mixed lymphocyte tumor culture (MLTC), using irradiated TRAMP-C tumor cells, were determined by flow cytometry to be a relatively even mix of CD4+ T cells and CD8+ T cells (not shown). To assay the cultured T cells for cytokine production, cells were harvested, isolated using density separation medium, and incubated in vitro with irradiated tumor cells for 24 hr as described in the methods section. Supernatants from 24 hr T cell cultures were assayed for IL-4, IL-17, TGF-ß1, IL- 10 and IFN-γ using specific cytokine captures ELISA’s. Only IL-10 and IFN-γ were detected (Figures 3A and 3B). The amount of IFN-γ present in 24 hr supernatants was significantly increased when T cells were cultured with relevant experimental targets, compared to control targets (Figure 3A). The amount of IFN-γ produced by T cells cultured with Yac-1 cells (MHC-I negative control) was neglible, and not detected for mKSA tumor cells (antigen positive, MHC-I mis-matched control). These data indicate that there are hD52-DNA vaccine-induced, tumor cell-specific CD8+ T cell responses in immunized mice. This was confirmed by the ability of H-2b, class I MHC specific mAb to inhibit production of IFN-γ when included in 24 hr cultures of T cells and TRAMP-C target cells (p < 0.05 for comparison of like cultures indicated by *) (Figure 3A). Control mAb failed to inhibit IFN-γ production by T cells cultured with TRAMP-C tumor cell targets (not shown). Taken together, these data suggest that hD52-DNA immunization induces an antigen-specific, class I MHC-restricted cellular immune response that is likely responsible for the observed tumor protection in vivo.
Though IL-10 was the only other cytokine detected in the supernatants from the 24 hr T cell cultures, the average amount of IL-10 produced was about 3-fold less than INF-γ. This suggests that IFN-γ played a more dominant role in tumor protection, and that in survivor mice, the vaccine was sufficient to induce immune responses capable of rejecting tumor challenge in the majority of mice. Similar to IFN-γ producing T cells, the IL-10 producing T cells were MHC-I restricted for all 5 animals tested (Figure 3B) as demonstrated by complete inhibition of IL-10 production in cultures with H-2b, class I MHC specific mAb. This suggests that IL-10 producing CD8+ Treg cells may be responsible for lack of complete protection in some immunized mice by suppressing effector CTLs via IL-10 mechanisms. Inhibition of IFN-γ production with specific mAb was only significant for 2/5 animals tested (Figure 3A). This may be due to the presence of CD4+ IFN-γ producing T cells in the cultures. It is interesting to speculate that the IL-10 production observed in MHC-I restricted T cells from protected mice may have been higher in nonprotected animals, suggesting CD8+ IL-10 producing suppressor cells were dominant. Since IL-4 and IL-17 were not detected, neither a Th2 nor a Th17 cellular immune response was elicited by hD52-DNA vaccination, nor did they play a role in tumor protection (summarized in Table 1). T cells cultured alone without tumor cells and tumor cells cultured alone without T cells failed to secrete detectable levels of any of the cytokines tested.
To assess CTL killing of tumor target cells, T cells from MLTC described above were subject to in vitro killing assays. Targets consisted of syngeneic MHC class-I matched TRAMP-C2 tumor cells (used for secondary tumor challenge), and MHC class-I mismatched, antigen- positive, mKSA tumor cells. The NK cell-specific target Yac-1 was also tested but not lysed, indicating absence of NK cells (not shown). CTLs generated from mice immunized i.m. with hD52-DNA that survived tumor challenge demonstrated tumorspecific lysis (Figure 3C), as TRAMP-C2 (H-2Kb) challenging tumor cells were lysed. The observed lytic values exceeded our set lower limit for the control H-2d target, mKSA (10%) (Figure 3C). Taken together, these data suggest that CTLs were generated following i.m. immunization with hD52-DNA, as demonstrated by greatly enhanced lysis of relevant target cells.
Overall tumor protection and T cell profiles induced by D52- DNA vaccination i.m. yielded similar effects. All four DNA vaccine strategies protected mice from primary TRAMP-C1 tumor challenge, as well as demonstrated memory protection to secondary TRAMP-C2 challenge. Only IL-10 and IFN-γ were detected, suggesting induction of Th1-type cellular immunity as well as CD8+ IL-10 producing suppressor T cells. This conclusion was supported by blocking of cytokine production with specific MHC class-I mAb (Table 1).
mD52-DNA vaccination with depletion of CD122hi T cells exacerbates tumor growth
We were interested in further studying the role CD8+ IL-10+ T cells might be playing in our D52 vaccine tumor models. Over the past decade the importance of CD4+ Treg cells in autoimmunity and tumor immunity has been well established. However, much less is known about suppressor T cells of the CD8+ subset. Given the importance of both CD4+ and CD8+ T effector cell subsets in complete and effective immunity, it is reasonable to argue that CD8+ Treg cells are equally important and involved in tolerance maintenance to over expressed tumor self proteins like D52. Recent studies suggest that CD8+ CD25hi (IL-2Ra chain) Foxp3+ Treg cells like CD4+ CD25hi Foxp3+ Treg cells inhibit immunity via cytokines other than IL-10 [30]. Conversely, CD8+ CD122hi (IL-2Rβ chain) Treg cells produce IL-10 to suppress CD8+ T cell effector function [31]. In light of these published studies along with our findings, we examined whether depletion of CD122hi (CD8+) T cells in conjunction with D52-DNA vaccination increased protection from tumor challenge, as has been demonstrated by others [32]. To this end, we immunized mice i.m. with mD52-DNA every two weeks for a total of four injections. mD52- DNA was chosen as the vaccine approach, because it was slightly less effective at protecting against tumor challenge compared to hD52, given us a larger window to see an effect from depletion of CD122hi CD8+ T cells. Depleting mAbs were administered i.p. with the first and third immunizations and at the time of tumor challenge 14 days after the last immunization. Groups of mice were either mock depleted, CD25 depleted (as a comparison for CD4 Treg function), CD122 depleted or depleted of both CD25hi and CD122hi T cells (Figure 4A). Our studies demonstrated that both mD52-DNA and hD52-DNA vaccines elicited tumor protective T cell responses and CD8+ IL-10+ T cells. We postulated that mD52-DNA vaccine, being 100% self compared to hD52, which is about 86% identical to mD52 at the amino acid level, may give us a better chance of eliciting CD8+ IL-10+ T cells and thus a better opportunity of determining if they are indeed CD122hi CD8+ Treg cells by depleting this subset in vivo. Peripheral blood lymphocytes from like mice in an experimental group were collected from the tail vein just prior to tumor challenge and pooled. Flow cytometry analysis of CD4 vs. CD25 and CD8 vs. CD122 expression on lymphocytes from immunized and control (mock) depleted mice compared to immunized and CD25 or CD122 depleted mice demonstrated that we were successful in obtaining approximately 70% depletion of the target cell populations (Figures 4B and 4C).
Analysis of s.c. TRAMP-C2 tumor growth over time revealed that mD52-DNA vaccination in the mock depleted mice was only capable of rejecting tumors in about 15% of the animals in this group (Figure 5A). This was comparable to what we observed in mD52-DNA immunized mice following secondary challenge with TRAMP-C2 (Figure 2B). This low level of protection was observed even with 50% reduction in the number of inoculated tumor cells (5 x 105) compared to our experiments in figure 2 (1 x 106 cells). TRAMP-C2 cells are nearly 5 times more tumorigenic than TRAMP-C1 cells [25]. To clearly test whether or not the CD8+ IL-10+ T cells were suppressive in function, it was necessary ensure maximum tumor growth, thus we challenged with TRAMP-C2 cells. Depletion of CD4+ CD25+ T cells (classic Treg cells) brought tumor protection in this stringent scenario back to about 50% (Figure 5B). This corroborated our findings in other murine tumor models that demonstrated an active role for classic CD4+ Treg cells in mD52 vaccine induced tumor immunity [27]. Importantly, neither the depletion of CD122hi T cells nor the simultaneous depletion of both CD25hi and CD122hi T cells with mD52-DNA vaccination increased primary tumor protection from TRAMP-C2 tumor cells (Figures 5C and 5D, respectively). Strikingly, the converse was observed, in that this approach exacerbated tumor growth in nearly 90% of the CD122 depleted mice and nearly 80% of the double depleted mice (Figure 5E). Since others have demonstrated an increase in autoimmunity in murine models when CD122hi T cells were depleted in a similar manner to our study using the same anti-CD122 mAb [33-35], our results indicate that the CD8+ IL- 10+ T cells elicited with D52-DNA vaccination cannot be classified as CD122hi CD8+ Treg cells, since depleting CD122hi T cells doesn’t augment tumor prevention by eliminating suppression to the tumor self antigen D52, but instead exacerbates tumor growth. Therefore, these CD8+ IL-10+ T cells may be a unique subset of CD8+ T cells distinct from CD122hi CD8+ regulatory T cells [33].
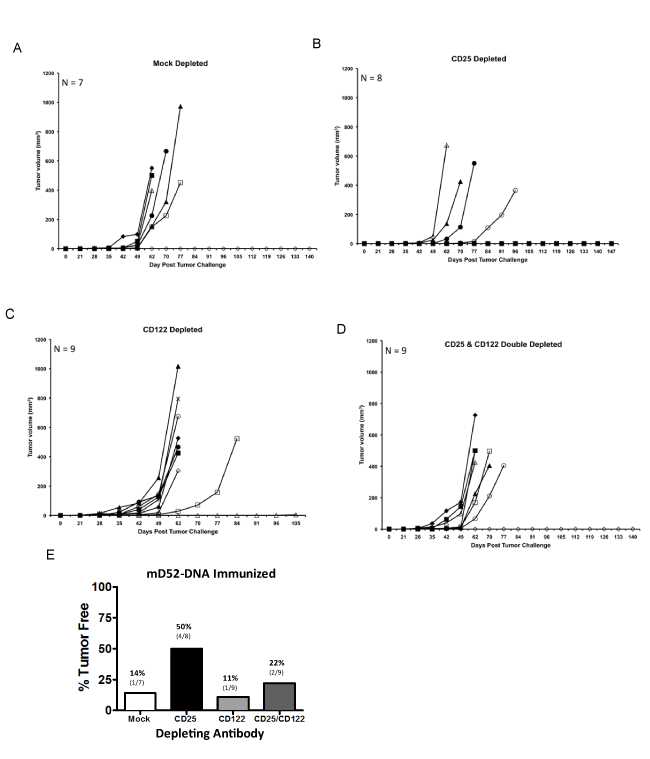
Figure 6: Relative expression of multiple immunologic genes in T cells from mD52 immunized mice. The X axis depicts expression of the genes shown relative to GAPDH for a given T cell population determined by realtime RT-PCR at 27 cycles. Upper panel A) histogram depicts representative flow cytometry data demonstrating that the cells analyzed were greater than 98% T cells. Lower panels (B, C) depict data from 2 representative mice selected from 10 mice that were immunized with mD52 DNA and challenged with TRAMP-C2 tumor cells. B) Bulk T cells. C) CD8+ T cells. ◊= T cells from immunized mice that were protected from tumor challenge. ♦= T cells from immunized mice that were not protected from tumor challenge. Values = mean +/- SEM for triplicates. Shown are representative data from two separate experiments.
D52-DNA vaccination elicits a subset of IL-10+ CD8+ T cells that are FOXP3 negative
To further explore the possibility that the CD8+ IL-10+ T cells are distinct from other well characterized regulatory T cells, we expanded (using CD3, CD28 magnetic beads as described in the methods section) and isolated pure T cells (98% CD3 positive) (Figure 6A) rom mD52-DNA immunized mice that rejected TRAMP-C2 tumor cell challenge and from immunized mice that delayed TRAMP-C2 tumor cell growth but eventually succumbed to the tumor. We compared expression of multiple T cell markers related to an effector or regulatory phenotype by real-time RT-PCR. Because some key markers of interest are not surface expressed proteins, and to increase the sensitivity of detection we chose to examine gene transcript levels using real time RT-PCR (Figure 6B). T cells from both groups of mice expressed high levels of CD3γ confirming our flow data. Interestingly, both groups expressed CD8a at levels near those of CD3γ, but T cells from tumor protected mice contained more CD4+ T cells than those from unprotected mice, albeit much less than CD8+ T cells. IL-10 transcript levels were higher in T cells from unprotected mice, in which IL-10 transcripts were not detected. Interestingly, T cells from both groups expressed high perforin and granzyme-b transcript levels. Notably, FOXP3 was not detected in T cells from either group. Isolated CD8+ T cells from both groups demonstrated that indeed CD8+ T cells were likely to be a source of the IL-10 being produced and that these CD8+ T cells were FOXP3 negative (Figure 6C). CD8 T cells from protected mice produced greater amounts of IFN-γ than T cells from mice that were not protected from tumor challenge, as illustrated in figure 3A. These data indicate that a FOXP3 negative subset of CD8+ T cells that produce IL-10 is induced D52-DNA vaccination. Together with our CD122 depletion data, these findings suggest that D52-vaccine immunity elicits a potentially unique subset of CD8+ T cells (not CD122hi and FOXP3 negative) that may be suppressive in function due to the preferential secretion of IL-10.
Conclusion
Our previous studies of vaccines targeting mD52 demonstrated protection against tumor challenge in mice when mD52 was administered as recombinant protein injected with CpG-ODN as a molecular adjuvant i.m. [24], or in conjunction with CD25 Treg depletion when protein was administered s.c. [27], or as a DNAbased vaccine administered s.c. with recombinant GM-CSF protein [26]. Of note, mD52 protein administered without a toll-like receptor (TLR)-9 agonist does not elicit an immune response, attesting to its self-nature and inherent immunologic tolerance. Others reported that an mD52 overlapping peptide vaccine was effective in a murine breast cancer model [36]. Three important facts were revealed by these early mD52-vaccine studies. First is the successful use of a simple vaccine formulation. Most vaccine studies to date pay little attention to the antigen and work to formulate vehicles for antigen delivery that themselves are immunogenic, as such are able to break olerance to the antigen being carried, yet a self-protein like mD52 is immunogenic when delivered as a simple protein, peptides or plasmid DNA as the vaccine formulation. Second is the critical demonstration that mD52-vaccines prevent tumor formation in mice without inducing autoimmunity [24], and unpublished observations. Third is the demonstration that inhibiting classic CD4+ CD25+ Treg cells with mD52 vaccination augments tumor immunity [27]. Together these studies demonstrate that D52 represents an over expressed tumor-self protein and a tumor vaccine candidate.
Herein, we tested the hypothesis that the xenogeneic human orthologue of D52 (hD52) when administered i.m. as a simple DNAbased vaccine would elicit an anti-tumor immune response that is more potent than that elicited by the murine othologue of D52 (D52) as assessed by protection from challenge with autochthonous TRAMP-C tumor cells which naturally contain elevated levels of mD52 protein [24]. TRAMP-C2 grows more aggressively than TRAMP-C1 in vivo and so was chosen as a more stringent test of vaccine durability to a recurrent tumor. In addition, TRAMP-C2 was originally derived from a distinct metastatic tumor so it may also represent a divergent recurrent tumor. Others have demonstrated that human xenogeneic tumor antigens are more immunogenic than the fully murine version of the antigens [37-40]. Our data corroborate these findings as the majority of mice (70%) that received four injections of hD52-DNA remained free from TRAMP-C1 tumor growth for nearly eight months and demonstrated delayed tumor onset (Figure 1B), compared to mice that received four injections of mD52 prior to tumor challenge (Figure 1C). Overall the protective tumor immunity induced by i.m. D52-DNA vaccination was considerable and did not vary greatly whether the vaccine was comprised of hD52-DNA, mD52-DNA or a combination of both (Figure 1). In addition, the DNA-based vaccines were durable over time as demonstrated by the ability to reject a second tumor challenge with TRAMP-C2 tumor cells administered more than four months after the initial tumor challenge (Figure 2). hD52- DNA induced protective tumor immunity was defined by a cellular immune response and the production of IFN-γ (Figure 3). Of note, the only other cytokine detected was IL-10 (Table 1), which appears to be of MHC-I-restricted, CD8+ T cell origin (Figure 3). This was consistent with observations from our previous mD52-based vaccine studies in two mouse strains [26,27]. Others have suggested that CD8+ T cells that secrete IL-10 in response to a self-antigen are CD122hi regulatory cells [41,32]. To test this with our tumor vaccine model, we depleted CD122hi T cells in conjuction with mD52-DNA vaccination,compared to depletion of CD25+ Treg cells and assessed protection from tumor challenge (Figure 4). We demonstrated previously that mD52 protein-based vaccination was augmented by concomitant depletion of CD25+ Treg cells [27]. We hypothesized that depletion of CD122hi CD8 T cells or depletion of both CD122hi CD8 T cells and CD25 Treg cells would augment tumor immunity. mD52-DNA was chosen as the vaccine approach because it was slightly less effective at protecting against tumor challenge compared to hD52, giving us a larger window to see an effect from depletion of CD122hi CD8+ T cells. Surprisingly, depletion of CD122hi CD8 T cells alone or in combination with CD25 Treg cells exacerbated tumor growth, whereas CD25 Treg depletion alone augemented tumor immunity (Figure 5). It is possible that all CD8 T cells including anti-tumor effectors were depleted thus leading to increased tumor growth, however others have reported targeted depletion of CD8+ CD122hi Treg cells using the same mAb we used and similar administration schedules. Thus, we believe our results indicate that the CD8+ IL- 10+ T cells induced by D52-vaccination are not the same as those reported by others, with respect to CD122hi expression. We were interested to determine whether these CD8+ IL-10+ T cells express detectable levels of FOXP3 transcripts, supporting their role as a T cell population with suppressor function in our system. When we interrogated purified T cells from immunized protected mice using real-time RT-PCR, we could not detect FOXP3 but did detect IL-10 as well as transcripts encoding proteins associated with T cell mediated killing, namely perforin and granzyme-b (Figure 6). Together our data demonstrate that D52-based vaccines not only elicit CD25hi classical Treg cells that can be down modulated by targeting with specific mAb resulting in increased tumor immunity, but also a potentially unique (not CD122hi, and not FOXP3+) subset of CD8+ T cells that may be involved in suppressing optimal vaccine induced tumor immunity.
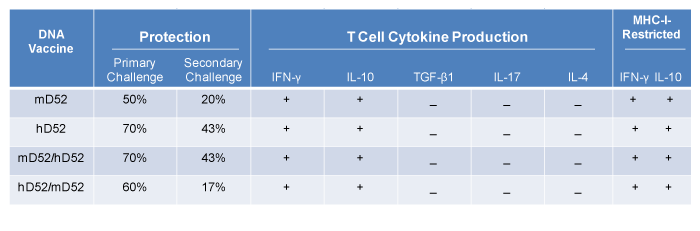
Table 1: Summary of D52 vaccine-induced tumor immunity and T cell cytokine profile. (Note:Shown are the results for primary TRAMP-C1 tumor challenge, secondary TRAMP-C2 tumor challenge and cytokine production from T cells co-cultured with TRAMP-C2 tumor cells. Splenocytes were harvested from surviving mice and co-cultured with irradiated tumor cells. Supernatants were collected after 24 hrs and assayed by ELISA for the production of IFN-γ, IL-10, TGF-β1, IL-17 and IL-4. T cells were co-cultured with mKSA and Yac-1 served as controls for nonspecific T-cell activity. In separate experiments, MHC-I H-2b blocking mAb was included with TRAMP-C2 tumor targets to assess MHC-I-Restriction.
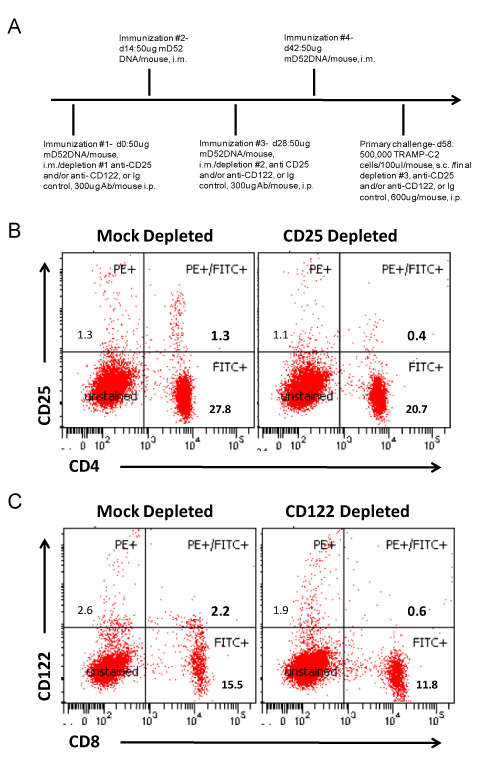
Figure 4: D52 DNA vaccination with depletion of CD25 and CD122 T cell subsets. Shown are representative data from male C57BL/6 mice immunized four times with mD52-DNA then TRAMP-C2 tumor challenged. A) A role for CD4+ CD25+ or CD8+ CD122+ regulatory T cells was assessed in response to mD52 DNA vaccination by in vivo depletion of the specific T cell subsetsat the time of the first and third vaccinations (300 μg per mouse, i.p.) and at the time of tumor challenge (600 μg per mouse, i.p.). Peripheral blood lymphocytes (PBLs) were collected just prior to s.c. tumor challenge with 5 x 105 TRAMP-C2 cells, and the lymphocytes isolated using Lympholyte-M density separation medium (Cedarlane Labs, Burlington, NC). Lymphocytes from animals in the same experimental group were pooled and stained with 1 μg each of anti-CD4-FITC and anti-CD25-PE mAbs or anti-CD8-FITC and anti-CD122-PE mAbs (BD Pharmingen) per 1 x 106 cells followed by analysis with a BD LSRII flow cytometer). Irrelevant IgG represented the mock depletion control. B) CD4, CD25 depletion analysis compared to mock depleted mice. C) CD8, CD122 depletion analysis compared to mock depleted mice. Representative dot plots from two separate experiments are shown.
Figure 6: Relative expression of multiple immunologic genes in T cells from mD52 immunized mice. The X axis depicts expression of the genes shown relative to GAPDH for a given T cell population determined by realtime RT-PCR at 27 cycles. Upper panel A) histogram depicts representative flow cytometry data demonstrating that the cells analyzed were greater than 98% T cells. Lower panels (B, C) depict data from 2 representative mice selected from 10 mice that were immunized with mD52 DNA and challenged with TRAMP-C2 tumor cells. B) Bulk T cells. C) CD8+ T cells. ◊= T cells from immunized mice that were protected from tumor challenge. ♦= T cells from immunized mice that were not protected from tumor challenge. Values = mean +/- SEM for triplicates. Shown are representative data from two separate experiments.
The CD8+ IL-10+ T cells elicited by our tumor-self antigen vaccine are neither CD122hi nor FOXP3+, suggesting they are distinct from CD8+ CD122hi Treg cells described in association with inflammatory bowel disease [42]. Is it possible that they are what others have described as Tc10 cells? A subset of CD8+ T cells that produce IL-10, coined Tc10 cells, has been described in association with preventing peripheral tissue damage at the site of active T cells responses against viral pathogens [43]. When elicited in response to controlling anti-viral immunity, Tc10 cells are believed to be a transient, reversible phenotype, not a divergent effector lineage [44], supporting the notion that CD8+ T cells that produce IL-10 may be effector cells, not suppressor cells [45]. Tc10 regulation of CD8+ T cell responses against viral infections is believed to result from a shift in CD8+ effector cells to Blimp-1 expression and IL-10 production, away from T-bet and IFN-γ [46]. Tc10 cells have been recently reported in association with relapsing-remitting multiple sclerosis as well [47]. In this clinical study it was concluded that in MS patients, Tc10 cells produce IFN-γ and IL-10 at the same time and may function to protect against brain damage. An argument supporting the notion that Tc10 cells are effectors that control aberrant immunity is the demonstration that they produce normal to high levels of perforin, granzyme-b and IFN-γ [44]. Similarly, CD8+ CD122hi IL-10+ Treg cells produce perforin and granzyme-b, but not IFN-γ, and may function to kill auto reactive T cells [42]. Similar observations that distinguish Tc10 cells from CD8+ CD122hi IL-10+ Treg cells are the co-expression of IFN-γ by Tc10 cells and CD122hi expression by CD8+ Treg cells.
Likewise our data show similarities to what has been described for Tc10 cells and CD8+ IL-10+ Treg cells, such as the absence of FOXP3 expression and the production of IL-10 by CD8+ T cells elicited by vaccination against an over-expressed tumor-self protein, with the expression of high levels of perforin and granzyme-b (Figures 3 and 6, Table 1). However, we don’t believe that CD122hi expression is a marker for these CD8+ T cells that produce IL-10, since in vivo depletion of CD122hi T cells did not augment tumor immunity as one would expect from other reports [32], but instead exacerbated tumor growth (Figures 4 and 5). Further, we don’t believe that CD8+ T cells are simultaneously producing both IFN-γ and IL-10 (Figure 3) as others have suggested for Tc10 cells [47]. We will address these apparent differences in more detail with future experiments employing IL-10 knockout mice and transfer of T cell subsets with D52 vaccination, as well as analysis of IFN-γ, IL-10, CD122 and Blimp-1 expression in T cells from D52 vaccinated mice. In addition, the CD8+ IL-10+ T cells elicited in our vaccine model are restricted by classical MHC-I (Figure 3) and therefore distinct from Qa-1-restricted CD8 Tregs [48]. Our data support the conclusion that these CD8+ IL-10+ T cells are potentially unique for the reasons stated, but also because our system is neither a model of autoimmunity nor a model of viral infection. Ours is a vaccine model against a ubiquitously expressed self-protein that is aberrantly over-expressed in numerous cancers. In this vaccine model CD8+ IL-10+ T cells are elicited in the absence of tumor or any disease state (tumor challenge coming after induction of immunity to D52 via vaccination), supporting the hypothesis that they may indeed represent a unique subset of CD8+ T cells that use IL-10 and apoptotic proteins to suppress T cell responses against self-proteins. Of note, we have observed these CD8+ IL-10+ T cells following D52 vaccination in more than one mouse strain, suggesting a conserved role for their function. It is also interesting that these CD8+ IL-10+ T cells are elicited by mD52 (fully self) vaccines and hD52 (partially xenogeneic) vaccines, suggesting that their epitope specificity may lie within the region of the D52 protein that is conserved between species.
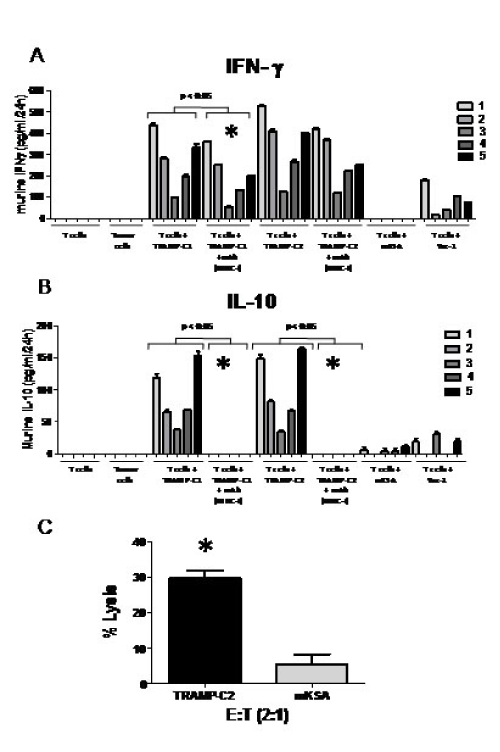
Figure 3: D52 DNA vaccine induces cell-mediated immunity and uncovers CD8+ IL-10+ T cells. Shown are representative data from male C57BL/6 mice immunized four times with hD52-DNA as described for figure 1. A and B) INF-γ and IL-10 cytokine production by T cells shown as picograms per ml of culture medium taken from 24h mixed lymphocyte tumor cells cultures. The shades of gray to black bars represent 5 individual mice in equal groups of 5, cultured alone or with various targets. Targets include: Autochthonous TRAMP-C1 and TRAMP-C2 tumor cells (mD52+, H-2b MHC-I-matched), mKSA tumor cells (mD52+, H-2d MHC-I-mis-matched) and Yac-1 tumor cells as an NK cell target control. To confirm MHC class I restriction, T cells were cultured with the challenging tumor cells in the presence of mAb specific for H-2b (T cells + TRAMP-C1 or C2 + mAb MCH-I). Values for T cells from individual mice cultured with tumor cells and an MHC-I blocking mAb are shown as mean +/- SEM for replicates. Data were analyzed using one-way ANOVA with Bonferoni multiple comparison post test. A p value of less than 0.05 was determined to be statistically significant (GraphPad Prism 5.0). Symbols (*) in graph 3A and B represent significant differences (p < 0.05) between like samples, indicating reduction in IFN-γ production or IL-10, respectively, in the presence of MHC-I blocking mAb. C) Bar graph showing specific lysis of TRAMP-C2 tumor cells at an effector-to-target cell (E: T) ratio of 2:1. The C57BL/6 mD52-expressing tumor cell line TRAMP-C2 served as an MHC-matched target. The Balb/c-derived mD52-expressing tumor cell line mKSA served as an MHC-mismatched control target. Values shown are the means ± SEM for triplicates and are representative of two repeat experiments. The symbol (*) represents significant differences (p < 0.05) in lysis between targets and was determined using one-way ANOVA and Tukey-Kramer Multiple Comparisons post test (GraphPad Prism 5.0).
In summary, the TRAMP model was employed to study DNAbased D52 vaccines against prostate cancer. Immunizations consisted of mD52-DNA, hD52-DNA or a combination of both, followed by challenge with mD52 positive, TRAMP-C1 tumor cells. Greater protection (70%) was observed 10 months post challenge in mice immunized with hD52 DNA. Survivors of the initial tumor challenge rejected a second tumor challenge with mD52 positive, autochthonous TRAMP-C2 tumor cells given in the opposite flank more than four months after the first challenge. Analysis of the T cell function from survivors indicated that a Th1-type cellular immune response was involved in tumor rejection. A potentially unique subset of CD8+ IL- 10+ T cells was also elicited and may play a role in inhibiting vaccine induced tumor immunity, suggesting that a deeper mechanistic understanding of these T cells in D52 vaccine-induced immunity may be important for developing a more potent cancer vaccine.
Acknowledgements
This work was supported by a grant from the DOD CDMRP PCRP: Award Number W81XWH-08-1-0660, and by funds from Texas Tech University Health Sciences Center.
References
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009; 15: 5323-5337.
- Schreiber TH, Raez L, Rosenblatt JD, Podack ER. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Sem Immunol. 2010; 22: 105-112.
- Shehata M, Weidenhofer J, Thamotharampillai K, Hardy JR and Byrne JA. Tumor protein D52 over expression and gene amplification in cancers from a mosaic of microarrays. Crit Rev Oncogenesis. 2008; 14: 33-55.
- Shehata M., Bieche I, Boutros R, Weidenhofer J, Fanayan S, et al. Non-redundant functions for tumor protein D52-like proteins support specific targeting of TPD52. Clin Ca Res. 2008; 14: 5050-5060.
- Lewis JD, Payton LA, Whitford JG, Byrne JA, Smith DI, et al. Induction of Tumorigenesis and Metastasis by the Murine Orthologue of Tumor Protein D52. Mol Cancer Res. 2007; 5: 133-144.
- Balleine RL, Fejzo MS, Sathasivam P, Basset P, Clarke CL, et al. The hD52 (TPD52) gene is a candidate target gene for events resulting in increased 8q21 copy number in human breast carcinoma. Genes Chromosom Cancer. 2000; 29: 48-57.
- Byrne JA, Tomasetto C, Garnier JM, Rouyer N, Mattei MG, et al. A Screening method to identify genes commonly overexpressed in carcinomas and the identification of a novel complementary DNA sequence. Cancer Res. 1995; 55: 2896-2903.
- Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A. 2002; 99: 12963-12968.
- Wang R, Xu J, Saramaki O, Visakorpi T, Sutherland WM, et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Res. 2004; 64: 1589-1594.
- Wang R, Xu J, Mabjeesh N, Zhu G, Zhou J, et al. PrLZ is expressed in normal prostate development and in human prostate cancer progression. Clin Cancer Res. 2007; 13: 6040-6048.
- Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, et al. Over expression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004; 64: 3814-3822.
- Byrne JA, Balleine RL, Fejzo MS, Mercieca J, Chiew YE, et al. Tumor protein D52 (TPD52) is over expressed and a gene amplification target in ovarian cancer. Int J Cancer. 2005; 117: 1049-1054.
- Largo C, Alvarez S, Saez B, Blesa D, Martin-Subero JI, et al. Identification of over expressed genes in frequently gained/amplified chromosome regions in multiple myeloma. Haematologica. 2006; 91: 184-191.
- Tiacci E, Orvietani PL, Bigerna B, Pucciarini A, Corthals GL, et al. Tumor protein D52 (TPD52): a novel B-cell/plasma-cell molecule with unique expression pattern and Ca(2+)-dependent association with annexin VI. Blood. 2005; 105: 2812-2820.
- Dave SS, Fu K, Wright GW, Lam LT, Kluin P, et al. Molecular diagnosis of Burkitts lymphoma. N Engl J Med. 2006; 354: 2431-2442.
- Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, et al. A bio logic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006; 354: 2419-2430.
- Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007; 98: 392-400.
- Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, et al. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006; 28: 315-326.
- Korkola JE, Heck S, Olshen AB, Reuter VE, Bosl GJ, et al. In vivo differentiation and genomic evolution in adult male germ cell tumors. Genes Chromosomes Cancer. 2008; 47: 43-55.
- McIntyre A, Summersgill B, Lu YJ, Missiaglia E, Kitazawa S, et al. Genomic copy number and expression patterns in testicular germ cell tumours. Br J Cancer. 2007; 97: 1707-1712.
- Hoek KS. DNA microarray analyses of melanoma gene expression: a decade in the mines. Pigment Cell Res. 2007; 20: 466-484.
- Roesch A, Becker B, Bentink S, Spang R, Vogl A, et al. Ataxia telangiectasia-mutated gene is a possible biomarker for discrimination of infiltrative deep penetrating nevi and metastatic vertical growth phase melanoma. Cancer Epidemiol Biomarkers Prev. 2007; 11: 2486-2490.
- Byrne JA, Mattei MG, Basset P. Definition of the D52 gene/protein family through cloning of D52 homologues in human (hD53) and mouse (mD52). Genomics. 1996; 35: 523-532.
- Payton LA, Lewis JD, Byrne JA, Bright RK. Vaccination with metastasis-related tumor associated antigen TPD52 and CpG/ODN induces protective tumor immunity. Cancer Immunol Immunother. 2008; 57: 799-811.
- Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997; 57: 3325-3330.
- Lewis JD, Sullivan LA, Byrne JA, de Riese W and Bright RK. Memory and cellular immunity induced by a DNA vaccine encoding self antigen TPD52 administered with soluble GM-CSF. Cancer Immunol Immunother. 2009; 58: 1337-1349.
- Bright JD, Schultz HN, Byrne JA, Bright RK. Injection site and regulatory T cells influence durable vaccine-induced tumor immunity to an over-expressed self tumor associated antigen. Oncoimmunology. 2013; 2: e25049.
- Lewis JD, Shearer MH, Kennedy RC, Bright RK. Surrogate Tumor Antigen Vaccination Induces Tumor-Specific Immunity and the Rejection of Spontaneous Metastases. Cancer Res 2005; 65: 2938-2946.
- Bright RK, Kimchi ET, Shearer MH, Kennedy RC, Pass HI. SV40 Tag-specific cytotoxic T lymphocytes generated from the peripheral blood of malignant pleural mesothelioma patients. Cancer Immunol Immunother 2002; 50: 682-690.
- Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, et al. CD8+ Foxp3+ regulatory T cells mediate immune suppression in prostate cancer. Clin Cancer Res. 2007; 13: 6947-6958.
- Endharti AT, Rifa'I M, Shi Z, Fukuoka Y, Nakahara Y, et al. CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol. 2005; 175: 7093-7097.
- Wang L-X, Li Y, Yang G, Pang P-Y, Haley D, et al. CD122+ CD8+ Treg suppress vaccine-induced antitumor immune responses in lymphodepleted mice. Eur J Immunol. 2010; 40: 1375-1385.
- Lee YH, Ishida Y, Rifa'i M, Shi Z, Isobe K, et al. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis.JImmunol.2008;180: 825-832.
- Saitoh O, Abiru N, Nakahara M, Nagayama Y. CD8+ CD122+ T cells, a newly identified regulatory T subset, negatively regulate grave’s hyperthydroidism in a murine model. Endocrinol. 2007; 148: 6040-6046.
- Motegi A, Kinoshita M, Inatsu A, Habu Y, Saitoh D, et al. IL-15-induced CD8+ CD122+ T cells increase antibacterial and anti-tumor immune responses: implications for immune function in aged mice. J Leukocyte Biol. 2008; 84: 1047-1056.
- Mirshahidi S, Kramer VG, Whitney JB, Essono S, Lee S, et al. Overlapping synthetic peptides encoding TPD52 as breast cancer vaccine in mice: prolonged survival. Vaccine. 2009; 27: 1825-1833.
- Gregor PD, Wolchok JD, Ferrone CR, Buchinshky H, Guevara-Patino JA, et al. CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine. 2004; 22: 1700-1708.
- Tormo D, Ferrer A, Bosch P, Gaffal E, Basner-Tschakarjan E, et al. Therapeutic efficacy of antigen-specific vaccination and toll-like receptor stimulation against established transplanted and autochthonous melanoma in mice. Cancer Res. 2006; 66: 5427-5435.
- Cheng-Fen T, Chi-Chen L, Ming-Chuan C, Tai-Ming K, Lin CM, et al. Autologous neu DNA vaccine can be as effective as xenogeneic neu DNA vaccine by altering administration route. Vaccine. 2007; 25: 719-728.
- Johnson LE, Frye TP and McNeel DM. Immunization with a prostate cancer xenoantigen elicits a xenoantigen epitope-specific T-cell response. OncoImmunol. 2012; 1: 1546-1556.
- Rifa'i M, Shi Z, Zhang SY, Lee YH, Shiku H, et al. CD8+ CD122+ regulatory T cells recognize activated T cells via conventional MHC class I-alpha betaTCR interaction and become IL-10-producing active regulatory cells. Int Immunol. 2008; 20: 937-947.
- Endharti AT, Okuno Y, Shi Z, Misawa N, Toyokuni S, et al. CD8+CD122+ regulatory T cells (Tregs) and CD4+ Tregs cooperatively prevent and cure CD4+ cell-induced colitis. J Immunol. 2011; 186: 41-52.
- Zhang N, Bevan MJ. CD8+ T cells: Foot soldiers of the immune system. Immunity. 2011; 35: 161-168.
- Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol. 2011; 186: 3642-3652.
- Mocellin S, Marincola FM, Young HA. Interleukin-10 and the immune response to cancer: a counterpoint. J Leuk Biol. 2005; 78: 1043-1051.
- Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1 dependent IL-10 production by anti-viral CTLs. Nat Immunol. 2011; 12: 327-334.
- Peelen E, Thewissen M, Knippenberg S, Smolders J, Muris AH, et al. Fraction of IL-10+ and IL-17+ CD8 T cells is increased in MS patients in remission and during a relapse, but is not influenced by immune modulators. J Neuroimmunol. 2013; 258: 77-84.
- Lu L, Cantor H. Generation and regulation of CD8+ regulatory T cells. Cell Mol Immunol. 2008; 5: 401-406.