
Case Report
Austin J Clin Med. 2014;1(2): 1007.
IgG4-Related Tubulointerstitial Nephritis Complicating the Course of Chronic Kidney Disease
Sun Y1, Luu NT1, Bijol V2, Vigil D1, Servilla KS1, Tzamaloukas AH1 and Konstantinov KN3*
Division of Nephrology, Raymond G. Murphy VA Medical Center and University of New Mexico School of Medicine, USA
Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, USA
Division of Rheumatology, Raymond G, Murphy VA Medical Center and University of New Mexico, USA
*Corresponding author: Konstantin N Kontantinov, Division of Rheumatology, Department of Internal Medicine, University of New Mexico School of Medicine, 2111 Lomas NE, 5 ACC Bldg.; Mail Stop MSC10-5550, Albuquerque, New Mexico 87131, USA
Received: February 02, 2014; Accepted: March 03, 2014; Published: March 06, 2014
Abstract
An 82-year old hadfor > 20 years chronic kidney disease (CKD) attributed to hypertensive nephrosclerosis. Proteinuria and abnormalities of the urine sediment had been consistently absent and the rise in serum creatinine had been slow, with levels < 1.80 mg/dL for several years. Then, serum creatinine rose to a peak of 2.83 mg/dL within three months, while urine findings remained unchanged and there were no clinical manifestations from other organs. Ultrasonographic study of the kidneys and urinary bladder was normal and volume replacement did not halt the progressive rise in serum creatinine. Serum IgG4 level was elevated and serum complement levels (both C3 and C4) were markedly depressed. Percutaneous kidney biopsy revealed both IgG4-related tubulointerstitial nephritis (TIN) and advanced chronic arterial and arteriolar sclerosis. Treatment with oral prednisone led to rapid improvement of serum creatinine concentrations, which returned to 1.80 mg/dL in one month, and of serologies with decrease in serum IgG4 concentration and sustained normalization of the serum complement levels. Development of IgG4-related TIN without radiographic or urinary changes in patients with preexisting CKD who had benign urinary sediment and no proteinuria, urinary tract obstruction, circulatory dysfunction or ingestion of nephrotoxic agents may lead to rapid deterioration of the renal function. In these patients, timely diagnosis guided by kidney biopsy leads to appropriate treatment which may reverse the rapid deterioration of the renal function.
Keywords: IgG4-related disease; IgG4-related tubulointerstitial nephritis; Hypertensive nephrosclerosis; Chronic kidney disease.
Abbreviations
IgG4-RD-IgG4-related disease; TIN-Tubulointerstitial Nephritis; MN-Membranous Nephropathy; CKD-Chronic Kidney Disease; CT-Computed Tomography; ANA-Antinuclear Antibodies; ANCA-Antineutrophil Cytoplasmic Antibodies.
Background
IgG4-related disease (IgG4-RD) is a recently characterized disease entitycomprising many previously described syndromes. The common histological characteristics of these syndromes are tumefactive lesions, dense lymphoplasmacytic infiltrates rich in IgG4-positive plasma cells, storiform fibrosis, and elevated serum IgG4 concentration [1-4]. The serological hallmarks of this entity, seen in the majority of the cases but not in every case, are serum levels of IgG4 above the normal range [5,6] and low serum complement levels. In one study with a small number of cases, serum complement levels reflected the level of activity of the disease, being low when the disease was active and returning to the normal range when the disease was inactive [7]. Criteria for the classification of a syndrome as IgG4- RD and for differentiation of other similar syndromes have recently been published [8-11]. The criteria proposed for the diagnosis of IgG4-RD are serum IgG4 concentration > 135 mg/dL, IgG4-positive plasma cells representing > 40% of the IgG-positive plasma cells in lesions, and >10 IgG4-positive plasma cells per high power field in biopsies of diseased organs, including the kidneys [11].
Patients with IgG4-RD have a predilection for forming mass lesions (pseudotumors) within organs, which can lead to misdiagnosis of cancer. IgG4-RD can also cause diffuse infiltrative lesions presenting without constitutional symptoms, which usually makes their diagnosis incidental and based upon radiographic finding or examination of pathological specimens. Spontaneous improvement is very rare, and the majority of cases show slow and indolent progression. Some patients have symptoms that overlap with allergic conditions, substantial elevation of serum IgE and peripheral eosinophilia. It is still unclear whether the disorder represents an autoimmune disease, chronic infection, or hypersensitive reaction. Familial cases are rare, and genetic studies of IgG4-RD are in their infancy. Small studies examining only Asian patients have implicated several HLA and non-HLA genes related to immune responses [13], but sufficiently powered search for genetic factors associated with susceptibility to known Th2 disorders and chronic inflammatory diseases has not been reported. Studies about bacterial infection as a triggering factor [14,15] and subsequent molecular mimicry underlie the anticipated importance of human microbiome for development of IgG4-RD.The contribution of innate immune responses inducing the production of large amounts of IgG4 [16] is also increasingly recognized.IgG4 production is controlled primarily by T helper 2 (Th2) cells and Th2-cell responses are predominantly activated at affected sites [17], possibly in conjunction with activation of regulatory T (Treg) cells.Autoreactive B cells and IgG4 autoantibodies play clearly an important role in IgG4-RD, but the antigen specificityfor the IgG4 antibody has not been defined. Potential autoantigens in glandular tissues include lactoferrin, carbonic anhydrases, and trypsinogens [18], but diagnostic and prognostic implications of serum IgG4 elevations at this point remain uncertain. For this reason, the diagnosis of IgG4-RD relies mainly on demonstration of characteristic histopathology and only secondarily on elevated serum IgG4/IgG ratio [18]. So far, there is no evidence to suggest that the IgG4-autoantibodies are directly pathogenic in IgG4-RD. This is in sharp contrast with certain immune-mediated conditions (pemphigus vulgaris and foliaceus, thrombotic thrombocytopenic purpura, and some cases of childhood membranous glomerulonephritis) in which autoreactive IgG4 autoantibodies induce direct damage by binding their cognate antigens [19].
Many IgG4 molecules in circulation consist of two different Fab arms making them “bi-specific” and functionally monovalent for a given antigen [20]. As a result, the circulatory IgG4 antibodies do not form immune complexes with antigens [20]. This makes them inefficient in phagocyte activation, antibody-dependent cellular cytotoxicity, andcomplement mediated damage. Interestingly, despite the fact that IgG4 seems to be unable to fix complement, hypocomplementemia is frequently observed especially in IgG4- related tubulointerstitial nephritis [21] and may precede worsening of the disease or improve with normalization of the serum complement [7].
The kidneys are one of the organs affected by IgG4-RD [21- 25]. The major histologicalform of IgG4-related renal disease is tubulointerstitial nephritis (TIN), also described as idiopathic hypocomplementemic TIN with extensive tubulointerstitial deposits [21-27]. IgG4-related TIN should be differentiated from granulomatosis with polyangiitis, Churg-Strauss syndrome, myeloma or lymphoproliferative disorders and rare forms of idiopathic hypocomplementemic TIN [28, 29]. IgG4-related TIN accounted for 27% of 44 cases of “primary” tubulointerstitial nephritis in one study [30] and 38% of 34 cases in another study [31].
The diagnosis of IgG4-related TIN is assisted by characteristic involvement in other organs, although the kidneys can be the only organ clinically involved, clinical features, serological findings and imaging techniques. Clinical features include abnormal kidney function and abnormal urinalysis. Serum creatinine level at the time of diagnosis varied between 0.67 and 6.87 mg/dL in a study of 23 patients [21] and between 0.9 and 9.0 mg/dL in a study of 35 patients [26]. In serological studies, elevated total IgG level was found in 100% [21] and 73% [26] of the cases, elevated IgG4 level in 100% [21] and 92% [26] of the cases, elevated serum IgE level in 57% [21] of the cases, hypocomplementemia in 70% [21] and 56% [26] of the cases, positiveantinuclear antibody [ANA] in 56% of the cases [21] and positive rheumatoid factor in 42% of the cases [21]. In another study of 16, patients with IgG4-related disease, elevated serum free κ light chain was found in 87% of the patients and elevated λ chains in 56%, while the κ:λ ratio was elevated in 44% of the cases [32]. Imaging abnormalities in the kidneys includemultiple small peripheral low-density lesions on contrast-enhanced computed tomography(CT), diffuse enlargement of the kidneys, hypertrophic lesions of the renal pelvic wall, and hypovascular solitary kidney masses [4,33]. Abnormal findings by imaging were detected in 70% [21] and 78% [26] of the patients with IgG4-related TIN.
The histological finding establishing the diagnosis of IgG4-related TIN is a large number of IgG4 positive plasma cells. The histological features often include storiform fibrosis and immune complex deposits in the tubular basement membrane. Interstitial fibrosis in kidneys of patients with IgG4-related TIN developing on the background of CKD can be the consequence of the primary renal disease causing the CKD. TIN is associated with IgG4-RD in other organs in the great majority of the cases [21]. IgG4-related TIN can also exhibit immune complex deposits in the basement membrane of the tubules, while the glomeruli are spared in cases without membranous nephropathy [22]. The histological characteristic which allows differentiation between IgG4-related and idiopathic hypocomplementemic TIN is the presence of plasma-cell rich interstitial infiltrate.
Membranous nephropathy (MN)is the second most common histological form of renal involvement in IgG4-RD [34-37]. IgG4- related MN is distinguished from the idiopathic variety by the fact the autoantibody involved in the pathogenesis of IgG4-related MN, although also an IgG4 molecule, is not directed against phospholipase A2 [34,36]. IgG4-related TIN and MN are found together in a number of patients [34,36]. Demographic and serological characteristics of patients with TIN only and those with TIN and MN are similar [38]. Histological forms of renal disease less frequently associated with IgG4- related disease include IgA nephropathy, membranoproliferative glomerulonephritis, endocapillary or mesangioproliferative immune complex glomerulonephritis and IgG4-related plasma cell arteritis of the renal vessels [23,39].
Sequential tests in patients with IgG4-related TIN show loss of renal function [4]. Urine findings are known to be important in the diagnosis of other parenchymal renal diseases. The urine abnormalities reported in patients with IgG4-related TIN include proteinuria, not in the nephrotic range unless there is also membranous nephropathy, and hematuria [4]. However, urine abnormalities are frequently absent. On repeated examinations of the urine in one series of 23 patients with TIN, 8 subjects (34%) had no proteinuria and another 7 subjects (30%) had proteinuria in only one of two urine specimens; in the same study, 15 subjects (65%) had no hematuria [21]. Pyuria and white cell casts, which are found in other varieties of interstitial nephritis, have not been reported. IgG4-related TIN, can lead to end-stage renal disease, but, unlike IgG4-related MN, responds usually to treatment with corticosteroids with improvement of the renal function despite the presence of interstitial fibrosis [40,41]. Several patients may have relapses, however [40,41].
The absence of characteristic urinary and imaging abnormalities complicates the diagnosis of IgG4-related TIN, especially when IgG4- related manifestations from other organ systems are missing [42]. We present a patient with IgG4-related TIN, absence of urine or imaging abnormalities and no extrarenal manifestations attributable to IgG4- related disease, who, in addition, carried for years the diagnosis of another renal disease causing chronic kidney disease (CKD) with no abnormalities in the urinalysis. This patient illustrates the need for aggressive diagnostic approach in patients with CKD, sudden deterioration of renal function, absence of proteinuria and bland urine sediment.
Case Presentation
An 82-year-old man underwent a percutaneous kidney biopsy. He had a history of hypertension for decades, radical prostatectomy 18 years in the past for prostatic carcinoma, Gleason’s stage 5, stable Parkinson’s disease for 12 years, gastro-esophageal reflux for many years and coronary artery disease with a coronary artery bypass graft 4 years in the past. Abdominal CT scan for epigastric pain was normal eight years prior to the admission for kidney biopsy. His serum creatinine levels were ≥ 1.3 mg/dL for >20 years and CKD had been formally diagnosed. In the five year following the diagnosis of CKD, his blood pressure was under control, serum creatinine concentration ranged between 1.36 and 1.74 mg/dL, without a clear-cut tendency to rise, he had no difficulty in urination, several ultrasonography studies showed consistently normal size and texture of the kidneys, no hydronephrosis and no residual urine in the bladder after urination, while numerous urinalysis examinations showed consistently absence of formed elements and determinations of the protein/creatinine ratio in spot urine specimens (normal value ≤ 0.2) were usually in the normal range with a couple of measurements at a ratio of 0.4. On the basis of these features CKD had been attributed to nephrosclerosis with probable contribution of bladder outlet obstruction prior to the prostatic surgery.
Serum creatinine concentration rose to values above 2.00 mg/dL two months prior to the kidney biopsy. Urinalysis and urine protein/ creatinine ratio, repeated many times during these two months, remained unremarkable, with the exception of one microscopic examination of the urine which revealed several granular casts and rare muddy brown casts, but no red cells or white cells. Ultrasonographic studies of the kidneys and urinary bladder were also unchanged. Infusion of saline in an attempt to correct a presumed extracellular volume deficit had no sustained effect on the rate of rise in serum creatinine.
At the time of admission for kidney biopsy, serum creatinine level was 2.83 mg/dL. His medications included carvidopa/levodopa, furosemide, metoprolol, omeprazole, simvastatin, tamsulocin, nitroglycerin and loratadine. Clinically, he had no fever, arthralgia, rash, edema, sialadenitis, lymphadenopathy, or dacryoadenitis. Chest x-ray disclosed no patchy infiltrates or nodular lesions. Serologic tests disclosed normal levels of ANA and ANA panel including anti-SS-A, anti-SS-B, anti-Sm, anti-DNA, and anti- RNP antibodies, antineutrophil cytoplasmic antibodies (ANCA), including myeloperoxidase-ANCA, and proteinase-3-ANCA, hepatitis antibody panel, cryoglobulin, and M-protein. Serum rheumatoid factor (normal range < 14 IU/mL), rose to a high of 29 IU/mL and returned within the normal range during treatment. Serum complement levels were depressed including a C3 value of 42 mg/dL (normal range 90-180 mg/dL) and a C4 value of < 3 mg/dL (normal range 16-47 mg/dL). Serum IgM level was slightly low (30 mg, dL, normal range 40-250 mg/dL), serum IgA was normal (202 mg/dL, normal range 82-453 mg/dL) while serum total IgG (2276 mg/dl, normal range 600-1630 mg/dL) and IgG4 levels (340 mg/dL, normal range 4-86 mg/dL) were elevated. Other abnormal serologic test included elevated values of serum IgE (497 IU/mL, normal <114 IU/mL) and of free light chains (free κ chain 173.6 mg/dL, normal range 3.3-19.4 mg/dL; free λ chain 112.9 mg/dL, normal range 5.7- 22.3 mg/dL). The κ:λ ratio (1.54) was in the normal range (0.26-1.65). The determinations of serum immunoglobulins, including IgG4 and of light chains were done after the results of the kidney biopsy became known.
Renal biopsy disclosed severe immune complex-mediated interstitial nephritis, with IgG4 positive plasma cell-rich infiltrate and tubular basement membrane deposits. Extensive infiltrate by mononuclear cells, with many plasma cells and occasional neutrophils was noted in the interstitium. The infiltrate involved the medulla diffusely and the cortex to a lesser extent. There was prominent invasion of the tubular epithelial cells by mononuclear inflammatory cells. Immunoperoxidase staining was performed for IgG, IgG4 and CD138 (plasma cell marker). The number of IgG4-positive plasma cells (> 30 per high power field) fulfilled the criteria for IgG4-related renal disease [11]. The glomeruli appeared normal on hematoxylin and eosin (H and E) stain and had slightly irregular basement membranes in thickness by Periodic Acid Schiff (PAS) staining. Advanced chronic changes of kidney parenchyma including tubular atrophy, advanced interstitial fibrosis (trichrome stain)without the pattern of storiform fibrosis, and severe arterial and arteriolar sclerosis were noted (Figures 1-5).
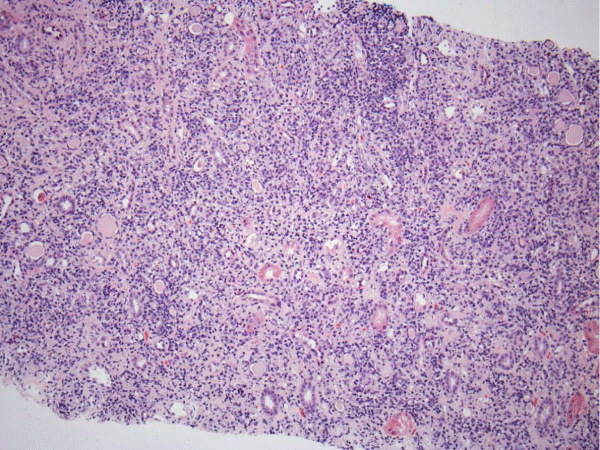
Figure 1: The interstitium appears diffusely infiltrated by inflammatory cells and many tubules appear atrophic, with a low epithelial lining and hyaline casts in their lumens (H&E, 100X).
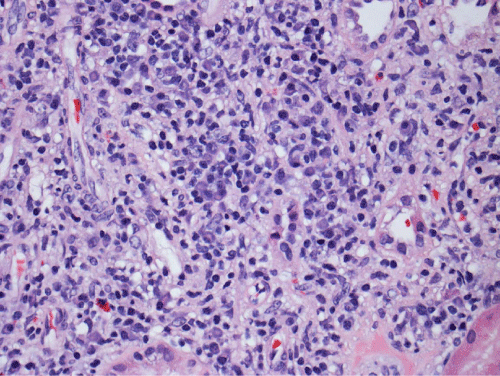
Figure 2: The inflammatory infiltrate is composed mainly of lymphocytes and many plasma cells that can be recognized by eccentric nuclei and basophilic cytoplasm (H&E, 200X).
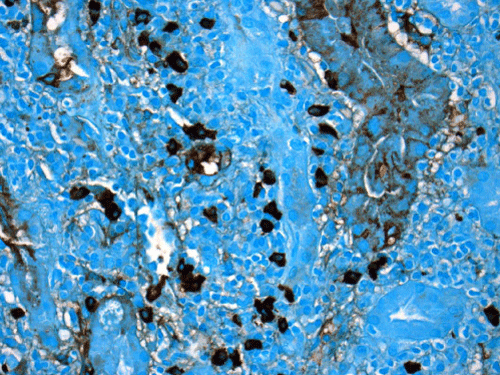
Figure 3: The immunoperoxidase staining for IgG4 is positive in many (>30) plasma cells on this high power magnification image (IgG4, 400X). Counterstain was done by methyl green/alcian blue mixture.
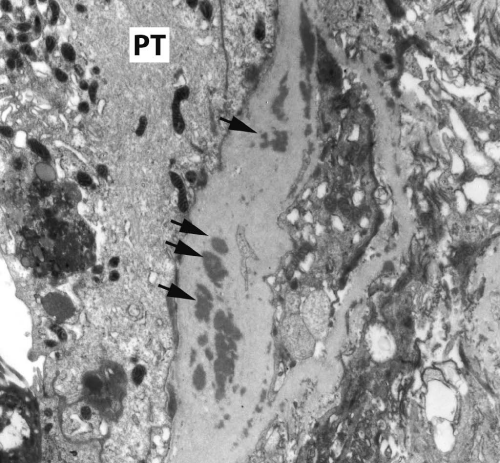
Figure 4: Tubular basement membranes contain several electron-dense deposits (Black arrows): to the left is the proximal tubular epithelium (PT) (electron micrograph, 10,000X).
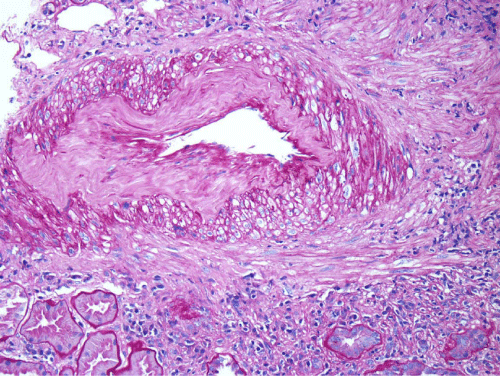
Figure 5: Tubular basement membranes contain several electron-dense deposits (Black arrows): to the left is the proximal tubular epithelium (PT) (electron micrograph, 10,000X).
The final histological diagnosis was IgG4-related TIN superimposed on chronic arteriosclerotic kidney disease. He was started on oral prednisone 40mg daily with a slow tapering to 5mg daily over two months. Serum creatinine improved from 2.83 to 1.91 mg/dL after two weeks and to 1.80mg/dL after six weeks of prednisone. Serum IgG4 level decreased from 340 to 165 mg/dL after two weeks of prednisone treatment. Serum total IgG returned to within the normal range in the same time period. Serum complement levels rose also promptly and returned to within the normal range one month after the start of treatment. They have remained in the normal range in five measurements. Urinalysis has remained clear throughout the treatment period. Urine protein/creatinine ratio was, in three consecutive determinations, 0.1, 0.4 and 0.0.
Conclusions
The only manifestation of IgG4-related TIN may be an elevation of serum creatinine, while extrarenal lesions of IgG-related disease, abnormal sonographic images of the kidneys, proteinuria, hematuria, and pyuria may be absent. IgG4-related TIN with these characteristics developing in the course of another condition causing CKD without abnormal urinary findings causes significant diagnostic difficulties. A definitive diagnosis can only be made by a kidney biopsy in this case. Kidney biopsy should be considered in patients, particularly elderly subjects, who during the course of CKD without proteinuria or hematuria attributed to nephrosenescence and nephrosclerosis develop a rapid and persistent worsening of the renal function. Recognition of IgG4-related TIN is critical for institution of appropriate therapy and salvage of renal function.
Consent
Consent for publication of this case was obtained from the patient and the human research review committee of this institution
Acknowledgements
This report was supported by the Raymond G. Murphy VA Medical Center.
References
- Divatia M, Kim SA, Ro JY. IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J. 2012; 53: 15-34.
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012; 366: 539-551.
- https://www.uptodate.com/contents/overview-of-igg4-related-disease?source=search_result&search=Overview+of+Ig4-related+disease&selectedTitle=1~150
- Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH . IgG4-Related Disease. Annu Rev Pathol. 2014; 9: 315-347.
- Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011; 23: 108-113.
- Yamamoto M, Tabeya T, Naishiro Y, Yajima H, Ishigami K. Value of serum IgG4 in the diagnosis of IgG4-related disease and in differentiation from rheumatic diseases and other diseases. Mod Rheumatol. 2012; 22: 419-425.
- Kihara M, Sugihara T, Hosoya T, Miyasaka N. Clinical significance of complement as a biomarker of disease activity in 4 cases of IgG4-related disease with retroperitoneal fibrosis. Clin Exp Rheumatol. 2013; 31: 947-949.
- Stone JH, Khrosroshahi A, Deshpande V, Chan JKC, Heathcote JG, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthr Rheumatism. 2012; 64: 3061-3067.
- Okazaki K, Umehara H. Are Classification Criteria for IgG4-RD Now Possible? The Concept of IgG4-Related Disease and Proposal of Comprehensive Diagnostic Criteria in Japan. Int J Rheumatol. 2012; 2012: 357071.
- Deshpande V, Zen Y, Chan JKC, Yi EE, Sato Y, et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathology. 2012; 25: 1181-1192.
- Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012; 22: 21-30.
- Zen Y, Nakanuma Y. Pathogenesis of IgG4-related disease. Curr Opin Rheumatol. 2011; 23: 114-118.
- Kountouras J, Zavos C, Chatzopoulos D. A concept on the role of Helicobacter pylori infection in autoimmune pancreatitis. J Cell Mol Med. 2005; 9: 196-207.
- Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009; 361: 2135-2142.
- Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012; 64: 914-924.
- Akitake R, Watanabe T, Zaima C, Uza N, Ida H. Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut. 2010; 59: 542-545.
- Lohr JM, Faissner R, Koczan D, Bewerunge P, Bassi C, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol. 2010; 105: 2060-2071.
- Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-Related Disease. Annu Rev Pathol. 2014; 9: 315-347.
- Guma M, Firestein GS. IgG4-related diseases. Best Pract Res Clin Rheumatol. 2012; 26: 425-438.
- Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002; 105: 9-19.
- Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010; 78:1016-1023.
- Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011; 15: 615-626.
- Cornell LD. IgG4-related kidney disease. Semin Diagn Pathol. 2012; 29: 245-250.
- Cornell LD. IgG4-related kidney disease. Curr Opin Nephrol Hypertens. 2012; 21: 279-288.
- Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int. 2014; 85: 251-257.
- Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC . Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011; 22: 1343-1352.
- Yamaguchi Y, Kanetsuna Y, Honda K, Yamanaka N, Kawano M, et al. Characteristic tubulointerstitial nephritis in IgG4-related disease. Human Pathology. 2012; 43: 536-539.
- Kambham N, Markowitz GS, Tanji N, Mansukhani MM, Orazi A. Idiopathic hypocomplementemic interstitial nephritis with extensive tubulointerstitial deposits. Am J Kidney Dis. 2001; 37: 388-399.
- Vaseemuddin M, Schwartz MM, Dunea G, Kraus MA. Idiopathic hypocomplementemic immune-complex-mediated tubulointerstitial nephritis. Nat Clin Pract Nephrol. 2007; 3: 50-58.
- Kim TY, Park KS, Choi JS, Kang SH, Cho YM. Comparative clinical manifestations of IgG4- related and IgG4-negative primary tubulointerstitial nephritis. Clin Nephrol. 2011; 76: 440-446.
- Yoshita K Kawano M, Mizushima I, Hara S, Ito Y, Imai N, et al. Light microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant. 2012; 27:2755-2761.
- Grados A, Ebbo M, Boucraut J, Vély F, Aucouturier P. Serum Immunoglobulin Free Light Chain Assessment in IgG4-Related Disease. Int J Rheumatol. 2013; 2013: 426759.
- Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007; 242: 791-801.
- Cravedi P, Abbate M, Gagliardini E, Galbusera M, Buelli S. Membranous nephropathy associated with IgG4-related disease. Am J Kidney Dis. 2011; 58: 272-275.
- Yahata M, Takahashi S, Nakaya I, Sakuma T, Sato H. Possible IgG4-related kidney disease requiring a differential diagnosis of membranous lupus nephritis. Intern Med. 2012; 51: 1731-1736.
- Alexander MP, Larsen CP, Gibson IW, Nasr SH, Sethi S. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013; 83: 455-462.
- Kanda H, Koya A, Uozaki H, Tateishi S, Sato K, et al. Membranous nephropathy with repeated flares in IgG4-related disease. Clin Kidney J. 2013; 0:1-4.
- Kawano M, Mizushima I, Yamaguchi Y, Imai N, Nishi S, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: Detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012; 609795.
- Sharma SG, Vlase HL, D'Agati VD. IgG4-related tubulointerstitial nephritis with plasma cell-rich renal arteritis. Am J Kidney Dis. 2013; 61: 638-643.
- Nishi S, Imai N, Yoshida K, Ito Y, Saeki T. Clinicopathological findings of immunoglobulin G4-related kidney disease. Clin Exp Nephrol. 2011; 15: 810-819.
- Wu Q, Nakazawa R, Tanaka H, Endoh M, Fukagawa M. A Retrospectively Diagnosed Case of IgG4-Related Tubulointerstitial Nephritis Showing Good Renal Outcome and Pathological Progress. Case Rep Nephrol. 2013; 2013: 953214.
- Sayed R, Cook T, Palmer A. Recognizing isolated IgG4-related nephropathy. Clin Kidney J. 2013; 0: 1-3.