
Review Article
Austin J Clin Neurol 2015;2(3): 1029.
Role of NLRP3 Inflammasome in Alzheimer’s Disease
Khan S1, Hashmet Parveen Ghouse1 and Fatima- Shad K2*
1Institute of Biomedical Sciences, University Brunei Darussalam, Brunei
2School of Medical and Molecular Biosciences, University of Technology Sydney (UTS), Australia
*Corresponding author: Fatima-Shad K, School of Medical and Molecular Biosciences, University of Technology, Sydney, 15 Broadway, NSW 2007, Australia
Received: March 15, 2015; Accepted: March 31, 2015; Published: April 09, 2015
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder in elderly. Amyloid beta (Aβ) aggregation and its association with specific receptors on microglia initiate chronic inflammatory response.
Nod like receptor family pyrin domain containing 3 gene (NLRP3) member of family of pattern recognition receptors (PRRs) initiate inflammation and apoptosis. NLPR3 inflammasome is a multiprotein complex and part of innate immune system. Injury induced activation of NLRP3 inflammasome via recognition of danger Associated molecular patterns (DAMPs) facilitate inflammasome complex formation and maturation of pro-IL-1β (and pro- IL-18) through caspases-1 and exacerbate the pathological condition by neuroinflammation. A review of the data shows that activation or inhibition of NLRP3 inflammasome leads to changes in process of neuroinflammation which may influence maturation and release of pro-IL-1β (and pro-IL-18) and Aβ accumulation and result in AD pathogenesis.
Keywords: Alzheimer’s disease; Amyloid β; NLPR3 inflammasome; Neuroinflammation; Microglia
Introduction
Alzheimer’s disease (AD) is the most common form of dementia and neurodegenerative disorder of elderly population [1]. The presence of two molecules extracellular Aβ in form of senile plaque and intracellular neurofibrillary tangle are characteristic feature of AD pathologies. In general, AD is associated with synaptic dysfunction, loss of neuronal circuits and networks [2]. However, the cause and subsequent development of pathologies of AD is still only partially understood, a number of genetic factors such as gene mutation, allelic variants all have been link to AD incidence. Furthermore, environmental factors also have influence on risk of AD development including exposure to metals their metabolism related to mitochondrial dysfunction, ROS production and apoptosis.
Both familiar and sporadic forms of AD share almost similar pathophysiology; Aβ accumulation, tau hyperphosphorylation and impaired axonal transport. These pathologic events cause toxic damage to cellular organelles resultant in ROS production and oxidative stress [3].
Aβ accumulation initiates an immune response by activating brain resident immune cells called “Microglia”. These microglias have specific receptors on their surface. Aβ peptide can activate these receptors. Activation of these receptors on microglia-mediate neuroinflammatory response [4,5]. Neuroinflammation is crucially associated with AD pathogenesis. It is further exacerbate the pathological condition and generate a plethora of inflammatory mediators and neurotoxic compounds. Inflammatory response in AD is initiated by Pattern recognition receptors (PRRs) (which are an integral part of immune system) recognize pathogen associate molecular patterns (PAMPs) and damage associated proteins (DAMPs) on glial cells, macrophages and oligodendrocytes within the brain. They can be membrane bound (toll-like receptors) or within the cytoplasm [Nod-like receptors (NLRs)]. NLRPs activation leads to assembly and activation of multiprotein complex known “inflammasome”. This inflammasome activation enables activation of pro-inflammatory caspases, particularly caspase-1. This then lead to activation and maturation of interleukin (IL)-1β, IL-18, and IL-33 [6,7].
Neuronal injury caused by insoluble Aβ peptide and tau tangle releases DAMPs which are recognized by PRRs (NLR domain) present in NLRP inflammasome initiate cascade of events leading to maturation and release of pro IL-1β, IL-18. Additionally, Aβ interaction with neuronal membrane cause efflux of K+ ions by forming ions channels, activates inflammasome which in turn secretion of cytokines. Purinergic P2X7 receptor activation decreases intracellular K+ levels. Impaired activity of Na+/K+ ATPase reduces ions gradients across the cell membrane cause cytotoxicity resulting neuronal cell death. Neuronal death serves danger signal by releasing DAMPs to activate NLRP3 inflammasome (Rubartelli, 2014). In addition with Aβ aggregation or protein misfolding and mitochondrial ROS give signal to NLRP3 inflammasome activation and up regulating pro inflammatory cytokines levels in brain resultant neuroinflammation [8].
Deficiency of NLRP3 inflammasome largely protects brain from deleterious effect of neuroinflammation. It is assuming that microglia-specific inflammasome as a promising cell type-specific molecular target in the CNS for therapeutic intervention for AD.
NLRs Structural Organization
There are 23 NLRs have been studied in human divided into four subfamily, Table 1 Most NLRs contain, nucleotide binding and oligomerization (NACHT) domain activates signaling pathways for proinflammatory cytokines. The C terminal contain leucine- rich repeat (LRRs) and an N terminal caspase and recruitment domain (CARD) or pyrin domain (PYD) as effector binding domain [9].
![]()
S/No.
NLR subgroup(s)
Protein(s)
Structure(s)
1
NLRA
CIITA(Class II Transactivator)
Acidic transactivation domain(AD)-N- terminal signaling domain-central nucleotide binding domain-C terminal sensing domain
2
NLRB
NAIPS(neuronal apoptosis inhibitor protein)
Baculovirus IAP repeat(BIP)-N- terminal signaling domain-central nucleotide binding domain-C terminal sensing domain
3
NLRC
NLRC4,NOD1,NOD2,NLRC3/C5/X1 Contain caspase recruitment domain
Caspase recruitment and activation domain (CARD)-N- terminal signaling domain-central nucleotide binding domain-C terminal sensing domain
4
NLRP
NLRP1,NLRP2-9,11-14,NLRP10 Contain pyrin domain
Pyrin domain(PYD)-N- terminal signaling domain-central nucleotide binding domain-C terminal sensing domain
Table 1: There are 23 NLRs in human have been studied which are subdivided into four sub-families NLRA or Class II, NLRBs, NAIPs, NLRCs and NLRPs a pyrin domain (PYD).
The PYD domain of NLRPs recruits the adaptor apoptosisassociated speck-like protein containing a caspase recruitment domain (ASC), which contains an N-terminal PYD and a C-terminal CARD. Additionally, the CARD domain of ASC represents an essential component for inflammasome formation via caspase-1 binding [9].
NLRP3 inflammasome structure
NLRP3 inflammasome is the most extensively studied among other inflammasomes. It is formed after oligomerization of NLRP3 and subsequent recruitment with apoptosis-associated Speck-like protein with a caspase recruitment domain (ASC) and procaspase-1. On activation this inflammasome forms large aggregate by assemble into fiber like structure that immensely amplifies the activation of caspase-1 [7] Figure 1.
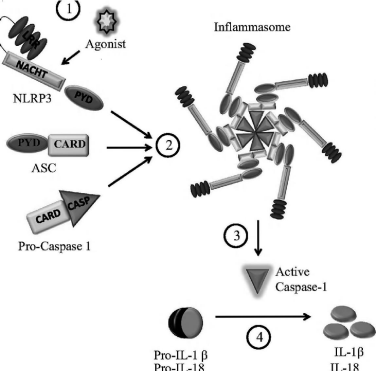
Figure 1: Adopted from [66]. Agonist stimulation of NLRP3 recruits
procaspase-1 through the adaptor protein ASC [apoptosis associated
speck like protein containing a caspase recruitment domain (CARD) to form
inflammasome, within inflammasome procaspase-1 undergo autocatalytic
processing, resulting in active caspase-1, which in turn cleaves pro IL-1β
into mature form.
NLRP3 inflammasome activation
Extracellular stimuli converge to activate NLRP3 inflammasome by mechanism including decrease intracellular concentration of K+, signaling via Ca2+, mitochondrial reactive oxygen species (ROS), Phagosomal destabilization and release of lysosomal cathepsins [10]. Calcium mobilization mediated mitochondrial damage in response to ATP can activate the NLRP3 inflammasome [11], and a purinergic signaling in particle mediated inflammation some activation pathways are also described [12] Figure 2.
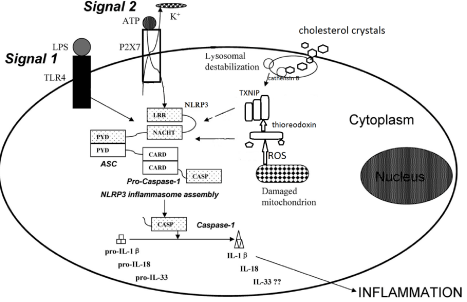
Figure 2: Adopted from [19], represent activation of NLRP3 inflammasome:
signal-1 via recognition of PAMPs through TLRs. Signal-2 extracellular ATP
serves ligand for ionotropic P2X purinoceptor 7 (P2X7) receptor leading to ion
fluxes such as K+ efflux through the P2X7 receptor. (I) NLRP3 activation via
direct sensing of DAMPs such as mitochondrial ROS-modified thioredoxin, the
thioredoxin-interacting protein (TXNIP) and (II) via indirect sensing of DAMPs
such as cholesterol crystals which may drive NLRP3 activation through
its internalization leading to phagolysosome destabilization associated
with leakage of cathepsin B allows inflammasome complex formation and
maturation of pro-IL-1β (and pro-IL-18) via caspases-1 cleavage to secretion
of bioactive IL-1β.
TLR-dependence
Toll like receptors (TLRs) belong to family of PRRs that regulate inflammasome activation via binding with ligands such as lipopolysaccharide (LPS). Nuclear factor-κB (NF-κB) consists of a family of transcription factors that are crucial for inflammation, immunity, and cell survival. NF-κB activation by TLRs leads to pro IL-1β secretion and IL-1β promoter activation [13]. In addition, TLRs facilitate NLRP3 inflammasome formation depended on NF-κB signaling representing a rate limiting component of inflammasome formation [14].
ATP-dependence
The NLRP3 inflammasome activation occur in response of infection or injury or to cellular stress including; increased extracellular ATP concentration, decreased in extracellular pH, cholesterol, monosodium urate crystals, and amyloid β aggregation [15,13].
P2X purinoreceptors 7 (P2X7R) ligand gated ion channel for ATP, found in microglia and macrophages. Cellular stimulation triggers ATP release and subsequent activation of P2X7R thereby regulating modulating cellular function in immunity. During pathologic condition ATP release from neurotoxic cells serves as danger signals activating NLRP3 inflammasome by binding through binding to the ionotropic P2X7 receptor [16].
ROS-dependence
Lysosomes are membrane bound cell organelles of spherical shape contain hydrolytic enzymes. They control intracellular turnover macromolecules. The presence of high content of hydrolytic enzymes makes lysosomes more harmful to cell. Lysosomal membrane damage reason to release their content into cytosol resultant degradation of cellular components and cell death. Lysosomal membrane permeabilization is one mechanism for induction of cell death. They are susceptible to Lysosomal membrane permeability inducing agents. In addition, production of intracellular ROS may induce Lysosomal permeabiltion only in those subcellular regions that are near to mitochondria, the major ROS-generating organelles. Additionally, chelatable iron and iron-catalyzed Fenton reactions, produce highly reactive pro-oxidants, and damage lysosomal membranes [17]. Lysosomal proteases are implicated for cell death is cathepsin such as cathepsin B (CB), cathepsin D (CD) and cathepsin L (CL) and, they can remain active at neutral ph. These cathepsins activate apoptotic effectors such as caspases and mitochondria. Caspase-1 activation via cathepsin B which suggests that the NLRP3 inflammasome recognizes lysosomal damage and subsequent lysosomal content in the cytoplasm following phagocytosis of NLRP3 agonists [18].
NLRP3 inflammasome in Alzheimer’s disease
It has been studied well that amyloid deposition cause cerebral neuroinflammation by activating microglia. In fact, Aβ is needed for NLRP3 inflammasome activation in microglia. It is fundamental for IL1-β maturation and subsequent inflammatory events in AD.
It is demonstrated that enhanced active caspase-1 expression in human with mild cognitive impairment and brain with AD suggested that NLRP3 inflammasome has a role in neurodegeneration in AD. Whereas, inhibition of NLRP3 largely protects brain from loss of spatial memory and decrease Aβ deposition seen in an AD mouse model. It also shows reduced caspase-1 and IL1-β activation as well as enhanced amyloid β clearance [20].
However, chronic deposition of Aβ gives signal to microglia activation that causes release of cytokines such as IL1-β [21]. Aβ light a fire in NLRP3 inflammasome and eventually induce AD pathology and tissue damage. Increased cleavage of caspase-1 was observed in AD patients’ brain mainly in the hippocampal area that consistent with chronic neuroinflammation [21]. Deficiency of NLRP3 inflammasome can recover neurobehavioral disturbance in AD has proven by scientists when used APP/PS1/ NLRP3-/- mice [20]. Amyloid precursor protein (APP) metabolism and Aβ aggregation and its clearance are also point of concern in AD associated neuroinflammation [22,23]. Increased number of microglia have seen around Aβ plaques showing that microglia are playing role in clearance of amyloid deposits by phagocytosis, as the AD progress the microglia adopted a chronically activated phenotype and cytokines deposition (IL1 β) disturb microglia clearance function [24,25]. NLRP3 inflammasome and caspase 1 activity decrease phagocytosis by microglia. It is suggested the amyloid-β-induced activation of the NLRP3 inflammasome enhances AD pathogenesis by producing inflammatory cytokines leading to synaptic dysfunction, cognitive impairment and beneficial microglia clearance of interleukins [20]. It is proven that NLRP3 or caspase-1 knockout mice show improvement in cognition due to suppression of amyloidosis and neuropathology Figure 3.
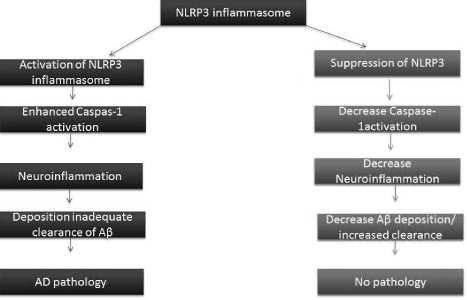
Figure 3: [26] Represent role of NLRP3 inflammasome in AD pathology
whereas, suppression with the NLRP3 inflammasome activation cause
decrease caspase-1 activity result in low amount of Aβ maturation. And
consequence of NO pathology.
NLRP3 inflammasome and Microglia in AD
Glia cells in the brain include; astrocyte, oligodendrocytes and microglia. Astrocytes provide balance normal internal environment, oligodendrocytes form, myelin sheath for neurons. Microglias are brain resident immune cells that provide defense system to brain. Microglia distribution density is higher in midbrain Substantia nigra compacta than other regions. It is noticed that midbrain region is more likely to be affected than other brain area in neurodegenerative diseases. It has shown that neuronal toxicity may depend on high distribution density of microglia. During the normal functioning of nervous system, microglias are in resting state. It is well noticed that microglia are very sensitive to external stimulation. This stimulation cause microglia reaction and is indicative of brain pathology. It is found that microglia has dual role in the development of AD i.e. nerve protective effect and immune damage. Excessive activation of microglia can cause neuron damage. In chronic condition microglia produce large amount of cytokines and ROS production [27]. Microglia activation involves receptor recognition which triggers assembly and activation of inflammasome. It is also responsible for caspases-1 mediated maturation of IL-1β activation [15]. IL-1β usually present at low levels in the healthy brain and its secretion could lead to several pathophysiological functions. IL-1β elevated level has been reported in cerebrospinal fluid and in postmortem brain tissues of patients with history of neuroinflammatory conditions as well as neurodegenerative disorders such as AD [28- 30]. Microglia activation is considered as early in AD that precedes the severe neutrophil destruction, clinical evidences proved presence of amyloid microglia complex in cerebral neocortex [31,32]. Lysosomal destabilization due to increased Aβ phagocytized by microglia can cause release of cathepsin B [33]. Released cathepsin B send signal to NLRP3 activation which further promote microglia synthesis and aggregation of inflammatory and neurotoxic factors [33,34].Microglia-specific activation of the NLRP3 inflammasome is pivotal for AD pathogenesis. NLRP3 inflammasome induces M1 like activation of microglia thus contribute to enhanced Aß deposition and cognitive impairment in AD reported in mouse model [35]. On the same time excessive secretion of IL-1β recruit to various members of the mitogen-activated protein kinase pathways [36]. It potentiates glutamate-induced neurotoxicity through the NMDA receptor [37,38] and involve in inducing iNOS in the hippocampus [39]. Toll like receptors expression on microglia have important role in pathological forms of inflammation and contribute to neurodegenerative disorders [40]. Whereas, purinoreceptors expression on microglia have ability to induce transcription of proinflammatory cytokines and take part in neuronal cell death [41] Figure 4.

Figure 4: Adopted from [42], represent the inflammatory response:
Recognition of damage signal lead to microglia activation and release of
proinflammatory cytokines. Constant exposure of brain immune cells to
damage signal causes more release of proinflammatory elements and
mutual activation of microglia cells along with proinflammatory cytokines
eventually trigger neuronal cell death. Dead neurons serve as danger signal
for neuroinflammation.
NLRP3 inflammasome and Mitochondria in AD
Hypermetabolism is the hallmark of AD and is associated with mitochondrial role in the neuropathology in AD [43]. It is critical for neurons function because the limited glycolytic capacity of these neurons make them highly dependent on aerobic oxidative phosphorylation for their energy needs. Mitochondria has also role in innate immunity. Mitochondria are considered important regulators of cytosolic homeostasis. It can sense and respond to changes in intracellular environment. Perturbation in intracellular K+, ROS, or lysosomal stability can result in mitochondrial dysfunction and apoptosis. It has been studied well that almost all aspect of mitochondrial functions are altered in Alzheimer’s neurons. Mitochondrial DNA (mt DNA) is particularly more vulnerable to oxidative damage then nuclear DNA in AD brain therefore, contribute to the neurodegenerative process. Mitochondrial dysfunction, abnormal mitochondrial dynamics contributing to its onset and progression when AD progress. Evidence from postmortem AD brains shows that alteration of several proteins involves in mitochondrial fission and fusion leading to abnormal redistribution of mitochondria. It has observed that mitochondrial dysfunction lead to increased oxidative stress and neurons dying by apoptosis in this way, mitochondria may trigger the abnormal onset of neuronal degeneration and death in AD [44]. Mitochondria are well positioned to regulate NLRP3 inflammasome. There are two signals in NLRP3 inflammasome activation; signal 1 often NF-kB activation, induces pro-IL-1β and NLRP3 expression. The second signal from any one of particulate matter, crystals, aggregated β-amyloid, extracellular ATP, or microbial toxins activates the NLRP3 inflammasome. However, reactive oxygen species (ROS) from mitochondria and mitochondrial DNA (mtDNA) are specifically responsible for activation of NLRP3 inflammasome [45]. Inhibitor of ROS can block priming step in NLRP3 activation [46]. There are several factors responsible for increased ROS production in mitochondria including various stress conditions, hypoxia, metabolic rate and membrane damage; increased ROS production leads to secretion of IL-1β reported in PC12 cells [47]. Mitochondria sense cellular danger that result in apoptosis, during which oxidized mtDNA is released into the cytosol. This mtDNA binds to NLRP3 and responsible for activation of NLRP3 and maturation of caspase-1 [48] Figure 5.
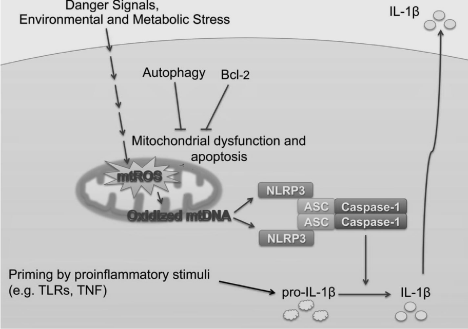
Figure 1: Adopted from [49], NLRP3 activators induce apoptosis that
activates the inflammasome ► Apoptosis is required for NLRP3 activation
► Oxidized mtDNA that is generated during apoptosis binds to NLRP3 and
activates it.
NLRP3 inflammasome and Endoplasmic reticulum in AD
The endoplasmic reticulum (ER) is specialized cellular organ that is necessary for protein folding and serves as site for biosynthesis for steroids, cholesterol and other lipids. It is responsible for cytosolic release of calcium [50]. ER stress in neurons has crucial role in pathologies in AD. ER can induce activation of several signaling pathways that are associated with unfold protein response (UPR) [51], that can trigger inflammatory responses as well as apoptotic cell death [52].
This UPR is responsible for maintaining ER homeostasis. During transient and mild ER stress, the UPR control ER homeostasis [53,54]. Whereas, at uncontrolled ER stress, UPR activates mitochondrial dependent or independent apoptotic pathways [55,53] via activation of NF-kβ [57,58]. ROS production through ER stress can activate ROS sensitive NLRP3 ligand Thioredoxin (TRX)-interacting protein (TXNIP). It dissociates from TRX and binds to NLRP3 upon excess ROS production to allow NLRP3 activation. This NLRP3 activation results in cleavage and secretion of IL-1β which indicative that ER stress serves as primer or activator for production of IL-1β and is involve in AD and other disorders [59,60]. On the same time, ER stress disturbs cellular Ca2+ homeostasis and activates inflammatory responses in host cells [61]. IP3 (Inositol triphosphate) gated Ca2+ channels have a key role in calcium regulation of neurons and perhaps involve in AD pathogenesis [62]. Phospholipase C catalysis and production of inositol 1-1, 4, 5trisphosphate cause release of Ca+2 from endoplasmic reticulum stores to extracellular environment[63,64] This increased in extracellular calcium demonstrated to provide feedback loop at a site of inflammation, thereby amplifying secretion of IL-1β and aggravating tissue damage [65], and play role in AD pathologies.
It is well noticed that neuronal-glia interaction is important for neuroinflammation as well as pathology of AD. In ER stress neurons are more vulnerable to than glia cells because UPRs is more pronounced in neurons than in glia cells. Moreover, the production of chemokines as alramin-type molecules in neurons due to ER stress affect the functions of glia cells (Salminen, et al., 2009). This is a logical way to prevent the over activation of microglia during stress conditions but this immunotolerance may impact detrimentally on microglial cleansing capacity and permit the aggregation of senile plaques.
Conclusion
The role of NLRP3 inflammasome in the pathogenesis of neurodegenerative diseases only recently being discovered and several levels of research are needed to fully understand the mechanisms of activation of NLRP3 inflammasome and its role in neuroinflammation and neurodegenerative disorders such as AD. There are many TLRs on cell surface and NLRs in the cytoplasm recognize danger signals (PAMPs and DAMPs) and initiate immune response. NLRP3 inflammasome activation is necessary for maturation of inflammatory cytokines/chemokines. Cellular components (Microglia, mitochondria and ER) express these receptors and participate in immunity. Prolong activation of NLRP3 inflammasome and release of cytokines/chemokines, and neuronal death serve as danger signal to NLRP3 activation, in this way neuronal cell death provide feedback loop and deteriorate the pathological condition. In addition, excess activation of glia cells trigger deleterious neuroinflammatory response. Moreover, prolong ER stress can be detrimental to neurons and can shift the UPR to switch on an apoptosis program. Mitochondria on the same time, can sense cellular danger that result in apoptosis, during which oxidized mtDNA is released into the cytosol. This mtDNA binds to NLRP3 and responsible for further activation of NLRP3 and maturation of caspase-1 and release cytokines. In conclusion all of these elements can aggravate the pathogenesis of AD. Deficiency or inhibition of NLRP3 inflammasome can be beneficial to reduce the deleterious effect of neuroinflammation in pathophysiology of AD. And an induction of therapeutic treatment target to NLRP3 inflammasome may be beneficial to AD patients.
References
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353-356.
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010; 13: 812-818.
- Matêj R, Rohan Z, Holada K, Olejár T. The contribution of proteinase-activated receptors to intracellular signaling, transcellular transport and autophagy in Alzheimer's disease. Curr Alzheimer Res. 2015; 12: 2-12.
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011; 14: 1227-1235.
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009; 64: 110-122.
- Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014; 8: 315.
- Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014; 15: 738-748.
- Rubartelli A. DAMP-Mediated Activation of NLRP3-Inflammasome in Brain Sterile Inflammation: The Fine Line between Healing and Neurodegeneration. Front Immunol. 2014; 5: 99.
- Choi AJ, Ryter SW. Inflammasomes: molecular regulation and implications for metabolic and cognitive diseases. Mol Cells. 2014; 37: 441-448.
- Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009; 227: 95-105.
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012; 109: 11282-11287.
- Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, et al. ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012; 3: e403.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140: 821-832.
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011; 186: 2529-2534.
- Lamkanfi M, Kanneganti TD, Franchi L, Núñez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. 2007; 82: 220-225.
- Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front inimmunol. 2012; 3: 414.
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008; 27: 6434-6451.
- Choi AJ, Ryter SW. Inflammasomes: molecular regulation and implications for metabolic and cognitive diseases. Mol Cells. 2014; 37: 441-448.
- Xie Q, Shen WW, Zhong J, Huang C, Zhang L, Li J. Lipopolysaccharide/adenosine triphosphate induces IL 1ß and IL-18 secretion through the NLRP3 inflammasome in RAW264. 7 murine macrophage cells. Int. J Mol Med. 2014; 34: 341-349.
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013; 493: 674-678.
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009; 27: 229-265.
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007; 184: 69-91.
- Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010; 117: 949-960.
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008; 28: 8354-8360.
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Nat. Acad. of Sci. 2010; 107: 6058-6063.
- Saco T, Parthasarathy PT, Cho Y, Lockey RF, Kolliputi N. Inflammasome: a new trigger of Alzheimer's disease. Front Aging Neurosci. 2014; 6: 80.
- Li Y, Tan MS, Jiang T, Tan L. Microglia in Alzheimer's disease. Biomed Res Int. 2014; 2014: 437483.
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005; 48: 16-42.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140: 918-934.
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010; 6: 193-201.
- Arends YM, Duyckaerts C, Rozemuller JM, Eikelenboom P, Hauw JJ. Microglia, amyloid and dementia in alzheimer disease. A correlative study. Neurobiol Aging. 2000; 21: 39-47.
- Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003; 24: 321-331.
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008; 9: 857-865.
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009; 27: 229-265.
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013; 493: 674-678.
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001; 21: 129-145.
- Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J Neuroinflammation. 2011; 8: 14.
- Ma XC, Gottschall PE, Chen LT, Wiranowska M, Phelps CP. Role and mechanisms of interleukin-1 in the modulation of neurotoxicity. Neuroimmunomodulation. 2002; 10: 199-207.
- Serou MJ, DeCoster MA, Bazan NG. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxidesynthase in primary hippocampal neuronal culture: plateletactivating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res. 1999; 58: 593–598.
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011; 472: 319-324.
- Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007; 446: 1091-1095.
- Maccioni RB, Far�as G, Morales I, Navarrete L. The revitalized tau hypothesis on Alzheimer's disease. Arch Med Res. 2010; 41: 226-231.
- Readnower RD, Sauerbeck AD, Sullivan PG. Mitochondria, Amyloid β, and Alzheimer's Disease. Int J Alzheimers Dis. 2011; 2011: 104545.
- Maruszak A, Zekanowski C. Mitochondrial dysfunction and Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2011; 35: 320-330.
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011; 12: 222-230.
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009; 183: 787-791.
- Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1b in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA. 1996; 93: 5635–5640.
- Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010; 22: 28-33.
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012; 36: 401-414.
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999; 13: 1211-1233.
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005; 74: 739-789.
- Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J Neuroinflammation. 2009; 6: 41.
- Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007; 32: 469-476.
- Land WG. Chronic allograft dysfunction: a model disorder of innate immunity. Biomed J. 2013; 36: 209-228.
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008; 7: 1013-1030.
- Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011; 23: 143-149.
- Kitamura M. Biphasic, bidirectional regulation of NF-kappaB by endoplasmic reticulum stress. Antioxid Redox Signal. 2009; 11: 2353-2364.
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004; 24: 1365-1377.
- Gotoh T, Endo M, Oike Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int J Inflam. 2011; 2011: 259462.
- Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005; 280: 847-851.
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005; 28: 37-43.
- Stutzmann GE. Calcium dysregulation, IP3 signaling, and Alzheimer's disease. Neuroscientist. 2005; 11: 110-115.
- Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003; 4: 530-538.
- Xi YH, Li HZ, Zhang WH, Wang LN, Zhang L, Lin Y, et al. The functional expression of calcium-sensing receptor in the differentiated THP-1 cells. Mol Cell Biochem. 2010; 342: 233-240.
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010; 6: 232-241.
- dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol. 2012; 303: L627-633.