
Case Report
Austin J Clin Neurol 2015;2(6): 1050.
Gliomatosis Cerebri in a Child: Another Case
Said NA¹, Waa S² and Samia P¹*
¹Department of Paediatrics and Child Health, The Aga Khan University, Kenya
²Department of Radiology, The Aga Khan University, Kenya
*Corresponding author: Samia P, Department of Paediatrics and Child Health, The Aga Khan University, 3rd Parklands Avenue, East Tower block, 5th floor No: 505, P.O.BOX: 30270, 0100, Nairobi, Kenya
Received: March 17, 2015; Accepted: May 16, 2015; Published: May 29, 2015
Abstract
Gliomatosis cerebri is a rare neuroepithelial tumor that is currently universally fatal. Its occurrence in the pediatric age group is uncommon and its diagnosis challenging. We present a case of Gliomatosis cerebri in a nine year old girl diagnosed through suggestive findings on neuroimaging and confirmed by histology of brain tissue obtained by stereotactic biopsy. We underline the need to have a high index of suspicion for GC in patients with recurrent seizures and suggestive neuroimaging findings.
Keywords: Gliomatosis cerebri; Brain tumor; Neuroimaging; Child; Seizures
Background
Gliomatosis cerebri (GC) is a rare form of diffusely infiltrating neuroepithelial tumor which is currently universally fatal. The World Health Organization (WHO) defines GC as a neuroepithelial neoplasm of unknown origin that involves at least two cortical lobes and may extend into infratentorial structures of the brain with preservation of the anatomic architecture and sparing of neurons [1]. It was first described in 1938 by Nevin [2]. The tumor may appear de novo (primary GC) or result from the spreading of a focal glioma (secondary GC). According to WHO criteria, primary GC can be classified into 2 types: Type 1 presents with diffuse invasion of cells without an obvious tumor mass, and Type 2 involves the diffuse invasion of cells beyond the lobe of a primary mass [1,3].
Since 1938, less than 300 cases of Gliomatosis cerebri have been described in literature with even fewer pediatric case reports [4]. Armstrong et al. reported the largest pediatric case series involving 13 patients followed up at the Children’s Hospital of Philadelphia [5]. Tailibert et al. reviewed 296 cases from literature and described an age range at presentation of 1 month to 85 years [4]. In the pediatric population, data supports a peak age at initial GC diagnosis to be in the second decade of life with the median age at diagnosis reported as 12 years. The median time to diagnosis from the first report of symptoms is three months (range, from 1 week to 20 months) [5].
Case Presentation
A nine year old girl, SB presented to the pediatric neurology service with a one month history of seizures. The first seizure was characterised by disorientation, staring and then eye blinking and unresponsiveness lasting approximately thirty seconds. Over the next two days she sustained five similar episodes. There was a positive history of postictal sleep. She had one episode of a generalized tonic clonic seizure lasting one minute followed by unresponsiveness and urine incontinence prompting a medical evaluation. EEG at that time was reported as normal. She was commenced on carbamazepine but she continued having many brief seizures every day.
SB was also reported to have new onset nocturnal enuresis and headaches. She had been observed to have reduced concentration and dropping school performance in the previous term.
Her birth history was unremarkable and there was no family history of convulsive disorder. Her subsequent development was reported as normal with no known chronic illness.
SB was well looking but rather quiet choosing to answer questions mostly in single words. Her Head circumference was growing just below the 50th centile, weight along the 25th centile and height along the 10th centile. She had a normal general examination with no neuro-cutaneous markers. SB’s cranial nerve and motor function examinations were normal apart from mild hypertonia and intermittent toe walking on the left side. No deformities were observed in her back.
A repeat EEG showed independent right temporal spikes, generalized poly-spike and wave activities and an asymmetric background, with the right side being relatively slower than the left. MRI brain was reported as having bilateral thalamic (predominantly right sided), right insular cortex and medial temporal lobe enlargement with signal abnormality of the caudate heads bilaterally (Figures 1,2,3).
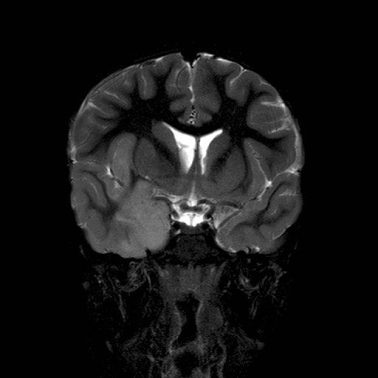
Figure 1: Coronal T2 weighted Image.
Demonstrates hyperintense and enlarged insular cortex and right medial
temporal lobe and effaced gyri.
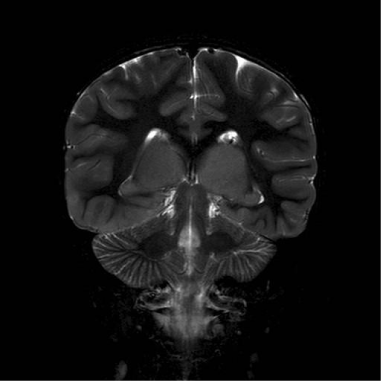
Figure 1: Coronal T2 weighted Image; shows bilaterally grossly enlarged and
hyperintense thalami. Basal ganglia are normal.
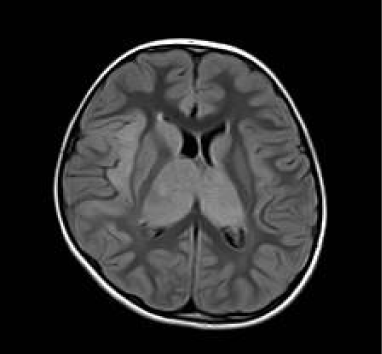
Figure 3: Axial Flair Image re-demonstrates enlarged hyperintense thalami
and right insular cortex.
SB was started on levetiracetam and carbamazepine weaned off without any further events for a month. She then presented with multiple seizure episodes characterised by confusion and incoherent speech followed by lethargy and sleep thereafter. She was admitted and received loading doses of Phenytoin and Sodium valproate. A third EEG during the admission showed intermittent delta and theta activities during the awake phase possibly due to postictal state. No spikes, poly spikes or sharp waves were noted. Sodium valproate was commenced and levetiracetam maintained.
Four months after onset of symptoms SB was noted to have brief daily seizures and a repeat MRI Brain was ordered. The MRI showed an extensive right medial temporal lesion, extending into enlarged caudate nuclei and both thalami suggestive of a diffuse neoplastic process. SB went on to have a stereotactic burr-hole procedure to obtain a biopsy of the lesion for definitive histological diagnosis. The histology was consistent with a WHO Grade II diffuse astrocytoma which, in view of the imaging features, confirmed a diagnosis of Type 1 Gliomatosis Cerebri.
The patient was started on treatment that included external beam radiotherapy 54Gy in 30 fractions and concurrent Temozolomide chemotherapy. Her anticonvulsants have been maintained as previously and she remains seizure free.
SB has just completed her radiotherapy course and is on a treatment break for four weeks before continuing on adjuvant chemotherapy. Repeat MRI Brain is planned eight weeks after completion of radiotherapy to serve as an interim assessment and baseline for future follow up.
Discussion
Age at presentation for SB does not fall far off the reported median of 12 years and given her residence in a developing country she is fortunate to have had biopsy confirmed diagnosis within four months of onset of symptoms. SB had seizures as the main presenting symptom. This is consistent with other reports documenting seizures as the main presenting symptom [6,7]. Jennings et al. reported the most common neurologic signs and symptoms in patients with GC which included corticospinal tract deficits (58%), dementia/ mental retardation (44%), headache (39%), seizures (38%), cranioneuropathies (37%), increased intracranial pressure (34%), and spinocerebellar deficits (33%). In the same study the authors reported three pediatric cases similar to SB, with intractable epilepsy as the presenting symptom of GC [6].
The diagnosis of GC is made using both neuroradiology and characteristic histopathologic findings on brain biopsy specimens [6,8,9]. Typically, MRI reveals diffuse, contiguous high-intensity areas on T2-weighted images and fluid-attenuated inversion recovery (FLAIR) images with variable contrast enhancement. On T1-weighted images GC is isointense to hypointense compared with normal brain. Enlargement of the brain lobes is well documented on T1-weighted images. According to Armstrong et al. in their report of the largest cohort of children with GC, the temporal lobe (54%), frontal lobe (38%), and parietal lobe (38%) were the most commonly involved sites. Involvement of the thalamus was also common. Our patient demonstrated these typical findings on MRI [5].
Despite advances in modern imaging modalities the diagnosis of GC remains a challenge [8]. Reports exist of patients having been diagnosed with ADEM or viral encephalitis before the diagnosis of GC was finally reached [10-12]. In our case the typical MRI Brain features correlated with the classic histopathology findings confirming the diagnosis.
References
- Fuller GN, Scheithauer BW. The 2007 Revised World Health Organization (WHO) Classification of Tumours of the Central Nervous System: newly codified entities. Brain Pathol. 2007; 17: 304-307.
- Nevin S. Gliomatosis cerebri. Brain. 1938; 64: 170-191.
- Sanson M, Napolitano M, Cartalat-Carel S, Taillibert S. [Gliomatosis cerebri]. Rev Neurol (Paris). 2005; 161: 173-181.
- Taillibert S, Chodkiewicz C, Laigle-Donadey F, Napolitano M, Cartalat-Carel S, Sanson M. Gliomatosis cerebri: a review of 296 cases from the ANOCEF database and the literature. J Neurooncol. 2006; 76: 201-205.
- Armstrong GT, Phillips PC, Rorke-Adams LB, Judkins AR, Localio AR, Fisher MJ. Gliomatosis cerebri: 20 years of experience at the Children's Hospital of Philadelphia. Cancer. 2006; 107: 1597-1606.
- Jennings MT, Frenchman M, Shehab T, Johnson MD, Creasy J, LaPorte K, et al. Gliomatosis cerebri presenting as intractable epilepsy during early childhood. J Child Neurol. 1995; 10: 37-45.
- Rajz GG, Nass D, Talianski E, Pfeffer R, Spiegelmann R, Cohen ZR. Presentation patterns and outcome of gliomatosis cerebri. Oncol Lett. 2012; 3: 209-213.https://www.ncbi.nlm.nih.gov/pubmed/16240166
- Caroli E, Orlando ER, Ferrante L. Gliomatosis cerebri in children. Case report and clinical considerations. Childs Nerv Syst. 2005; 21: 1000-1003.
- Qian R, Wu M, Wei X, Ling S, Ji Y, Fu X, et al. [Neuroimaging diagnosis and therapeutic efficacy of different surgical methods of gliomatosis cerebri]. Zhonghua Yi Xue Za Zhi. 2014; 94: 1639-1642.
- Kamaleshwaran KK, Krishnan V, Mohanan V, Shibu D, Shinto AS. Gliomatosis cerebri mimicking encephalitis evaluated using fluorine-18 fluorodeoxyglucose: Positron emission tomography/computed tomography. Indian J Nucl Med. 2015; 30: 84-85.
- McCusker MW, McDonald MA, Looby S, King M. Gliomatosis cerebri mimicking viral encephalitis in a 4-year-old child. BMJ Case Rep. 2014; 2014.
- Richard HT, Harrison JF, Abel TW, Maertens P, Martino AM, Sosnowski JS. Pediatric gliomatosis cerebri mimicking acute disseminated encephalomyelitis. Pediatrics. 2010; 126: e479-482.