
Case Report
Austin J Clin Neurol 2015;2(8): 1067.
Fatal Coma in a Young Adult due to Late-Onset Urea Cycle Deficiency Presenting with a Prolonged Seizure: A Case Report
Majid Alameri, Mustafa Shakra and Taoufik Alsaadi*
Department of Neurology Sheikh Khalifa Medical City (SKMC), Abu Dhabi, UAE
*Corresponding author: Taoufik Alsaadi, Department of Neurology, Sheikh Khalifa Medical City (SKMC), P.O. Box 51900. Abu Dhabi, United Arab Emirates
Received: April 23, 2015; Accepted: June 22, 2015; Published: June 30, 2015
Abstract
Ornithine transcarbamylase (OTC) deficiency is a urea cycle X-linked defect which considered the most prevalent inherited defect of the urea cycle. A very few reports of late-onset presentations in adults exist. Here, we report a previously healthy 17-year-old male who developed a prolonged seizure, a rapidly decline inmental status leading to coma over 3 days period. Analysis of the OTC gene showed a 119G variant, which was identified in exon 2 of the OTC gene by sequencing. A diagnosis of OTC deficiency should be considered in adult patients with unexplained hyperammonemic coma. This report highlights the pathophysiologic characteristics of this raredisorder, with this extremely rare presentation, clinical course, diagnosis and reviewthe therapeutic options.
Keywords: Ornithine transcarbamylase deficiency (OTC); Hyperammonemia; Coma
Introduction
Ornithine transcarbamylase (OTC) deficiency is a urea cycle X-linked defect, which is considered the most prevalent inherited defect of the urea cycle [1]. While typical presentation occurs in infancy, a few reported cases of late-onset presentations in adults are well described [2]. However, the delayed presentation is most commonly seen in partial OTC deficiency [2]. Males are usually more commonly, and most severely affected, than females [3]. The presentation vary, according to the degree of X-chromosome inactivation, from mild symptoms of fatigue and lethargy, to severe encephalopathy, coma leading to death [4].
The urea cycle is the primary metabolic pathway for the excretion of nitrogenous wastes such as urea. OTC catalyzes the mitochondrial reaction of ornithine (the end product of the extraction of urea from arginine) with carbamoyl phosphate (the first storage form of ammonia) to produce citrulline. Deficiency of OTC leads to the formation of excess carbamoyl phosphate; some of which is excreted as an orotic acid. When this pathway is overwhelmed, hyperammonemia results. Thus, patients with OTC deficiency end to have hyperammonemia, elevated levels of orotic acid in the urine, and low plasma citrulline [5].
Males and heterozygous females with partial OTC deficiency can present from infancy to adulthood. One observational study of 21 male patients found the age at presentation ranges from 2 months to 44 years [6]. Furthermore, it was observed that male patients who were older at presentation had a diverse form of presenting symptoms and were associated with higher mortality rates [6]. This data illustrates the phenotypic variability of OTC deficiency even in the mild form of the disease (partial OTC deficiency), where a hyperammonemic crisis can be precipitated easily, and become a life-threatening condition very rapidly. For all individuals with OTC deficiency, a wide spectrum of neuropsychological complications have been described, including developmental delay, intellectual disability, attention deficit hyperactivity disorder (ADHD), and executive function deficits [7].
Here, we report a previously healthy 17-year-old male who developed intermittentnausea and vomiting for 1 week, followed by a witnessed new onset prolonged generalizedtonic-clonic seizure, rapid deepening coma over a 3 days period. A week prior to presentation, he started to take high protein supplement. Investigations for liver disease, drug and alcohol use, and infections were all negative. Blood NH3 concentration was remarkably high on initial presentation, but started to decrease with medical therapy and dialysis. During recovery, the patient developed Ventilator-associated pneumonia, treated with antibiotics, but unfortunately the course was further complicated by sever clostridium difficile colitis, miliaria profunda, progressing rapidly resulting in patient’s death. The diagnosisof OTC deficiency was confirmed by genetic analysis showing a 119G variant which was identified in exon 2 of the OTC gene by sequencing.
Case Presentation
A 17-year-old male previously healthy was admitted following a new onset generalized tonic-clonic seizure. The seizure was witnessed by his mother. She reported a continuous convulsion lasting for almost 6 minutes associated with urinary incontinence. Prior to admission, he had generalized body fatigue, intermittent nausea and vomiting for one week, without any associated features, such as fever, chills, diarrhea, abdominal pain, chest pain, shortness of breath, and weight loss. He was diagnosed as gastritis in a primary health care clinic, and was treated symptomatically. Upon further inquiry, here ported recently joining a local fitness club and starting on high protein supplement for one week prior to the development of the presenting symptoms.
On admission, he was afebrile with normal vital signs. The neurologic examination revealed that he was intermittently arousal to painful stimuli. His Glasgow Coma Scale score was 10 (E4, V2, M4), with the reminder of the exam being non focal. Initial laboratory data showed normal leukocyte count 10.7 × 109/L (N 4.5–13 × 109/L), normal glucose, electrolytes and calcium level. His liver function tests were as follows: Alkaline phosphatase: 179 IU/L (N 40-129 IU/L) , AST = 40 IU/L (N <40 IU/L), ALT =166 IU/L (< 41 IU/L), total bilirubin = 8 microm/L (≤17 microm/L), an ammonia level of 787 micromol/L (N16–60 umol/L) with normal coagulation panel. His initial chest X-ray, CT scan of the brain, and MRI/MRA of the brain were all normal. His initial prolonged EEG showed diffuse background slowing, without any evidence of electro graphical seizures, or epileptic form discharges
An Ultrasound scan of abdomen and pelvis with liver Doppler showed normal hepatic parenchyma and patent hepatic vessels without any other abnormality. Over the first 24 hour of admission, he became less responsive and his ammonia level failed to normalize despite lactulose therapy. He was intubated for airway protection. A repeat CT of the brain was done on the second day of critical care admission, showing diffuse effacement of sulcal spaces with decreased attenuation of the cerebral parenchyma, suggesting mild diffuse cerebral edema (Figure 2). In view of persistently elevated serum ammonia levels and brain edema, dialysis was initiated, plasma and urine amino acid analysis and urine organic acid quantitation were performed. Blood test showed Low level of citrulline (7 μM; normal 19-62 μM) and the urine test revealed elevated levels of orotic acid (27.7 mmol/mol creatinine; normal 0- 1.3 mmol/mol creatinine). Based on clinical and laboratory findings, aurea cycle disorder was strongly suspected and the patient was started on ammonia scavenger therapy arginine, sodium benzoate with intermittent hemodialysis. After the third hemodialysis session along with the medical therapy, the patient’s ammonia levels significantly, decreased over the course of treatment (median 55, average 184μmol/L, range 30–787μmol/L) (Figure 1), without any observable improvement in his level of consciousness.
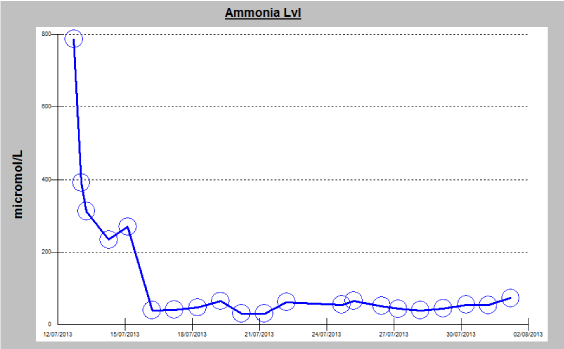
Figure 1: Diagram showing a timeline of the measured ammonia level in the
serum, observation of an exceptionally elevated level of 787 umol/L (normal
range = 11–35 umol/L) can be noted on the initial presentation, responding
to medical therapy with gradual decline during hospital stay (ammonia
scavenger therapy arginine, sodium benzoate and intermittent hemodialysis).
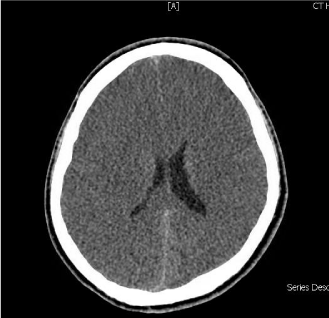
Figure 2: CT brain without contrast showing a generalized loss of the
supratentorial grey-white matter differentiation with effacement of sulci
indicating increasing degree of diffuse cerebral edema. Asymmetry of lateral
ventricles is seen with slit like appearance of anterior horns. An effacement of
the basal cisterns can be noted as well.
At that time, a repeat EEG showed very severely attenuated, poorly organized, and non-reactive EEG that was observed at high sensitivity, indicating a profound generalized disturbance of cerebral activity. He was kept on maintenance medical therapy and intermittent hemodialysis. His ammonia level was subsequently maintained within normal range without improvement in the level of consciousness failing multiple ventilation weaning trials. During the recovery period, he developed Ventilator-associated pneumonia treated with intravenous Piperacillin/tazobactam for 10 days, but unfortunately, the course was further complicated by sever clostridium difficile colitis, miliaria profundaended with cardiopulmonary arrest and death despite aggressive restitution.
The initial genetic testing for the OTC deficiency did not show any deletion and no duplication was identified in the OTC gene by Multiplex ligation-dependent probe amplification (MLPA) analysis. However, and because of the strong clinical suspicion, this result was not considered as conclusive, as it could not exclude OTC variants, where any variants outside of the analyzed region might not be detectable by MLPA and still might be present.Sequence analysis of the OTC gene was recommended and the diagnosis was confirmed showing 119G variant which was identified in exon 2 of the OTC gene. Based on these results, all of the patient siblings were recommended to be screened and were referredfor genetic consulting.
Discussion
Adult onset OTC deficiency is a rare cause of encephalopathy. The persistent elevated ammonia level may be the first clue to the diagnosis. Most of the adult onset patients remain asymptomatic, till they present with rapid decline in mental status, either spontaneously, or following, a heavy protein challenge, as the case in our patient [8]. Our case, similar to the previously few reported cases of OTC deficiency, present with rapidly worsening come that unfortunately can be potentially fatal [9]. Indeed, death is a usual outcome of the disease in its mild forms. However in severe cases, death may occur, either because of liver failure, or due to, similar to our patient, intercurrent severe illness and fatal inflammatory response [10]. On the other hand, seizures have been reported rarely as the first manifestation of OTC deficiency. Our patient had a reported prolonged seizure, unlike generalized convulsive seizures, which typically last less than 2 minutes. We realize the limitation of depending on witnesses to estimate the duration of a convulsive seizure; however, due to the concern of missing a non-convulsive seizure following the reported prolonged seizure, a 6- hour continuous EEG was obtained ruling out that possibility.
The basis of treatment of the acute status of hyperammonic coma is to target the plasma ammonia level to ≤200 μmol/through the use of ammonia scavengers treatment to allow excretion of excess nitrogen, which is expected to reduce the risk of neurologic damage, and if needed, a renal replacement therapy can further aid to lower the ammonia levels. Our patient’s ammonia level has responded nicely to treatment but his mental status did not. This lack of correlation between declining ammonia levels andthe improvement in mental status is quite puzzling. As a matter of fact, a recently reported series of 5 adult-onset OTC deficiency have demonstrated similar finding, where 3 of their patients had a normal ammonia levels days after initiation of treatment, but their cognitive status have not improved leading to death [11].
Liver transplant is typically considered in those who have frequent hyper ammonic episodes [12]. After acute phase, routine measurement of plasma ammonia, amino acids, liver function every three to six months may be required [7].
The diagnosis of OTCD in adults may be difficult, as clinical manifestations are non-specific, and often times, episodic in nature, which add more difficulties in considering the diagnosis. Moreover, laboratory findings can be normal outside the acute phase. Molecular analysis is the method of choice for confirming an OTCD.
Conclusion
Urea cycle disorders are one of the complex medical metabolic emergencies that occur in infancy till the adulthood period and must be treated promptly to avoid severe brain injury and death. However, there is very limited number of case reports in the literature for cases presenting in young adults. This should call on all physicians to consider this diagnosis for any adults presenting with unexplained hyperammonic coma and unexplained change in mental status. In these cases, every effort should be made for rapid screening of this condition, and for immediate treatment along with precise genetic counseling. Future efforts should look into widening the knowledge about the combined effect of genetic factors (i.e. mutations in the OTC gene, variants in modifier genes or epigenetic features) and environmental conditions in determining the phenotypic expression of OTCD.
References
- Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000; 105: e10.
- Cavicchi C, Donati M, Parini R, Rigoldi M, Bernardi M, Orfei F, et al. Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment. Orphanet J Rare Dis. 2014; 9: 105.
- GH T. High male: female ratio of germ-line mutations: an alternative explanation for postulated gestational lethality in males in X-linked dominant disorder. 2015; 58: 1364-1368.
- Yorifuji T, Muroi J, Uematsu A, Tanaka K, Kiwaki K, Endo F, et al. X-inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clin Genet. 1998; 54: 349-353.
- Milner JA, Visek WJ. Urinary metabolites characteristic of urea-cycle amino acid deficiency. Metabolism. 1975; 24: 643-651.
- Finkelstein JE, Hauser ER, Leonard CO, Brusilow SW. Late-onset ornithine transcarbamylase deficiency in male patients. J Pediatr. 1990; 117: 897-902.
- Lichter-Konecki U, Caldovic L, Morizono H and Simpson K. Ornithine Transcarbamylase Deficiency. University of Washington, Seattle. 2013.
- Spada M, Guardamagna O, Rabier D, van der Meer S, Parvy P, Bardet J, et al. (1994). Recurrent episodes of bizarre behavior in a boy with ornithine transcarbamylase deficiency: Diagnostic failure of protein loading and allopurinol challenge tests. The Journal of Pediatrics. 1994; 125: 249-251.
- Klein OD, Kostiner DR, Weisiger K, Moffatt E, Lindeman N, Goodman S, et al. Acute fatal presentation of ornithine transcarbamylase deficiency in a previously healthy male. Hepatol Int. 2008; 2: 390-394.
- Raper S, Chirmule N, Lee F, Wivel N, Bagg A, Gao G, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Molecular Genetics and Metabolism. 2003; 80: 148-158.
- Cavicchi C, Donati M, Parini R, Rigoldi M, Bernardi M, Orfei F, et al. Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment. Orphanet J Rare Dis. 2014; 9: 105.
- Busuttil AA, Goss JA, Seu P, Dulkanchainun TS, Yanni GS, McDiarmid SV, et al. The role of orthotopic liver transplantation in the treatment of ornithine transcarbamylase deficiency. Liver Transpl Surg. 1998; 4: 350-354.