
Research Article
Austin J Clin Ophthalmol. 2015; 2(4): 1057.
Histopathology of the Retina from a Three Year-Old Suspected to Have Joubert Syndrome
Bonilha VL1,2*, Rayborn ME¹, Bell BA¹, Marino MJ¹, Traboulsi EI¹, Hagstrom SA1,2 and Hollyfield JG1,2
¹Cole Eye Institute, Cleveland Clinic, Cleveland, USA
²Department of Ophthalmology, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USA
*Corresponding author: Bonilha VL, Cole Eye Institute, Cleveland Clinic, Ophthalmic Research - i3, 19500 Euclid Avenue, Cleveland, OH 44195, USA
Received: June 23, 2015; Accepted: August 25, 2015; Published: September 21, 2015
Abstract
Purpose: To define the retinal pathology in a 3 year-old eye donor who died from complications of an undiagnosed genetic syndrome.
Methods: Eyes were fixed and analyzed using macroscopic fundus photography (MF), confocal scanning laser ophthalmoscopy (cSLO) and spectral-domain optical coherence tomography (SD-OCT). Small areas from the perifovea and periphery were processed for histology and indirect immunofluorescence, using antibodies specific to retinal proteins such as rhodopsin, cone arrestin, RPE65 and others. Available medical records were also reviewed.
Results: With all three imaging modalities, the affected donor’s eyes lacked the distinct morphological detail typically observed with these techniques in postmortem control eyes. MF images showed a “photonegative effect” due to a hypopigmented macula relative to a hyperpigmented retinal background. cSLO imaging demonstrated a weak autofluorescence signal that was largely devoid of the usual retinal structures compared to the control. SD-OCT suggested disorganization of the affected retina, absence of a photoreceptor layer, and degeneration of the choroid in the macular area. Histologic findings indicated a highly disorganized photoreceptor layer in the macula and periphery. The RPE layer displayed thinning in some regions of the periphery and decreased pigmentation in most areas. Rods and cones were significantly reduced in the affected retina but a few cones were detected in the perifovea. Centrin-2 labeling was mostly absent from the connecting cilium of the photoreceptor cells. Medical record review pointed to a possible clinical diagnosis of Joubert syndrome.
Conclusions: The retinal degenerative findings, and absence of centrin-2 labeling are compatible with the expected retinal phenotype in patients with Joubert syndrome.
Keywords: Genetic abnormalities; Retina; Histology; Disorganized photoreceptors; Immunohistochemistry
Introduction
Genetic testing has allowed the identification of the underlying cause of a large number of inherited eye diseases. Eye conditions such as coloboma, microphthalmia, and the retinal dystrophies can be isolated, or can be part of a clinical syndrome [1]. It has been well established that there is significant genetic, allelic, and phenotypic heterogeneity amongst genetic eye diseases, especially the inherited retinal dystrophies. While clinical presentation and examination can often provide a clinical diagnosis, genetic testing and the demonstration of the underlying molecular pathology establish the cause of disease.
Here we report the analysis of eyes of a male donor with profound visual impairment. The patient had received a clinical diagnosis of Leber Congenital Amaurosis (LCA). Genetic testing was carried out but failed to identify the disease-causing mutation(s). A medical record review indicated a wider range of clinical signs and symptoms and suggested the possibility of a syndromic form of retinal dystrophy.
Materials and Methods
Patient information
The organ donation was coordinated through The Foundation Fighting Blindness Rare Eye Donor Program (#852). The review of medical records and tissue analysis were performed with the approval of the Cleveland Clinic Institutional Review Board (IRB #14-057). This male child was the product of a consanguineous relationship between his mother and her maternal uncle. He was removed from his mother and placed in foster care. The donor died at the age of 3 years from respiratory distress. When his globes were donated to this program, he carried a clinical diagnosis of Leber congenital amaurosis (LCA).
Genetic analysis
Peripheral blood was collected from the donor. DNA was extracted and purified from leukocytes in the blood by means of the Gentra Systems PUREGENE DNA Purification Kit (Qiagen). Since the presenting diagnosis was LCA, DNA was evaluated by Asper Ophthalmics for 641 mutations in 13 genes known to cause LCA (AIPL1, CRB1, CRX, GUCY2D, LRAT, MERTK, CEP290, RDH12, RPGRIP1, RPE65, TULP1, LCA5, SPATA7). However, due to insufficient DNA quality, some amplicons were missed and the analysis failed to identify any specific genetic mutation.
Medical Record Review
Approximately 5 years after obtaining this eye donation, the donor’s medical records were obtained through consent and coordination with the Washington State Department of Social and Health Services. Both the genetic counselor and ophthalmic geneticist analyzed all records. Based on the review of the donor’s clinical findings, a differential diagnosis was developed.
Ex-vivo Imaging of globes
Macroscopic fundus images of the posterior globes were acquired as previously described [2]. Briefly, the globes were bisected along the equator, and the posterior poles were transferred to a custommade plexiglass chamber filled with PBS solution to a level above the cut surface of the globe, and the posterior poles were imaged using a Zeiss AxioCam MRC5 camera and an angled illumination from a bifurcated fiber optic light source.
Scanning Laser Ophthalmoscopy (SLO)
SLO images of the fixed posterior poles were collected using a model HRA2 confocal scanning laser ophthalmoscope (Heidelberg Engineering, Inc.) as previously described [2]. The HRA2 was rotated 90o and the system was operated in high-resolution mode, which provides an image pixel format of 1536 x 1536 when used with a 55° wide-field objective lens. SLO images of the posterior pole were collected using Infrared Reflectance (SLO-IR), and Autofluorescence (SLO-AF) imaging modes at field of view (FOV) settings of 55°, 35°, and 25°.
Spectral Domain Optical Coherence Tomography (SD-OCT)
SD-OCT images of the posterior pole were collected using an SDOCT system (Model SDOIS, Bioptigen, Inc.), as previously described [2]. SD-OCT imaging was performed using the following scan parameters: (1) 5mm linear scan of the horizontal meridian through the optic nerve and fovea @ 1000 A-scans/B-scan, (2) 10mm linear scan of the horizontal meridian through the optic nerve and fovea @ 1000 A-scans/B-scan, (3) 5mm² Volume Scan of the posterior pole @ 500 B-Scans/Volume x 250 A-scans/B-scan, and (4) 10mm² volume scan of the posterior pole @ 500 B-Scans/Volume x 250 A-scans/Bscan. Post-acquisition images were averaged with a line and frame filter settings of 3 and 3, respectively using the Bioptigen InVivoVue SDOIS Software version 1.7.0.1645.
Histopathology
The globes were fixed 11 hours post-mortem in a mixture of 4% paraformaldehyde and 0.5% glutaraldehyde made in PBS. After 1 month in fixative, the globes were transferred and stored in 2% paraformaldehyde prepared in the same buffer. Eyes from a 3 year-old female with no history of retinal disease, who died from asphyxiation, were used as controls and were fixed 2 hours post-mortem. A small area of the retina/RPE/choroid tissue from the perifovea and periphery of the affected donor and age-matched control were cut and further processed as previously described [2]. Cryosections (8μm) were cut and labeled with the following antibodies: B630N to rhodopsin (1:50, from Dr. G. Adamus, Oregon Health and Science University, Portland, OR), 7G6 to cone arrestin (1:100, from Dr. P. MacLeish, Morehouse School of Medicine, Atlanta, GA), ab10062 to GFAP (1:400, Abcam), PETLET to RPE65 (1:500, from Dr. R. Crouch, University of South Carolina, Charleston, SC), AB1778 to calbindin D (1:500, Chemicon) and sc-27794 to centrin-2 (1:50, Santa Cruz). Cell nuclei were labeled with TO-PRO®-3 iodide (1mg/ml, Molecular Probes, Eugene, OR). Secondary antibodies (goat anti-mouse or anti-rabbit IgG; 1:1000) were labeled with Alexa Fluor 488 (green; Molecular Probes) and Alexa Fluor 594 (red; Molecular Probes). Sections were analyzed using the same acquisition parameters in a Leica laser scanning confocal microscope (TCS-SP2, Leica, Exton, PA). A series of 1 μm xy en face sections were collected through the whole section and processed into a three-dimensional projection of the entire cryosection (sum of all images in the stack). Microscopic panels were composed using AdobePhotoshop CS3 (Adobe, San Jose, CA).
Results
When first examined at the age of 1.5 years, the donor was found to have profound visual impairment and was diagnosed with LCA. He had failure to thrive (less than 3rd percentile for weight), hypotonia, developmental delay (one assessment showed 2 - 5 months developmental level at chronological age of 16 months), microcephaly (less than 3rd percentile), plagiocephaly, hyperextensible joints, pectus excavatum, and an undescended left testicle. An MRI of the brain showed mild to moderate cerebellar atrophy, with a hypoplastic inferior cerebellar vermis, and dilation of the fourth ventricle. A hearing test was normal. Additional exams included normal chromosomal analysis, negative for fragile X syndrome, negative for Smith- Lemli- Opitz syndrome, normal very long chain fatty acids, and normal extended newborn screen. The donor sustained several episodes of aspiration pneumonia as well as numerous other medical problems such as respiratory obstruction, hypertension, hypotension, and bradycardia. He underwent surgical interventions including tonsillectomy and adenoidectomy, fundoplication, gastrojejunostomy, and right orchiopexy.
As we characterized the retinal histopathology of this donor we intended to determine the specific mutation affecting this donor. However, there was insufficient DNA to perform additional genetic testing.
Fundus imaging of the affected donor eye showed a “photonegative effect” in the macula due to a hypopigmented macula relative to a hyperpigmented, dark retinal background (Figure 1B, arrow) when compared to the control eye (Figure 1A). SLO-IR imaging identified the optic disk (Figure 1D, asterisk) and the hypopigmented macula identified by fundus image (Figure 1D, arrow). SLO-AF imaging identified a weak AF signal (Figure 1F) that was devoid of any structural detail compared to the control, which clearly showed retinal vasculature and lipofuscin AF background (Figure 1E). All imaging modalities identified precipitated crystals from the buffer in the fixative solution in the control eye (Figures 1A, 1C, 1E, arrowheads).

Figure 1: Atrophic maculopathy in affected donor eye. A-B: Fundus image
of affected donor (B) and age-matched control eye (A). C-D: SLO-IR imaging
of affected donor (D) and matched control eye (C). E-F: SLO-AF imaging
of affected donor (F) and matched control eye (E). White arrows show the
atrophic maculopathy, white arrowheads show the precipitated crystals from
the buffer in the fixative solution, and asterisks show optic nerve head. Bar
= 0.2 cm.
SD-OCT en face view displayed a remarkable lack of structural features in the retina of the affected donor eye (Figure 2B) when compared to control eye (Figure 2A), which clearly showed the optic nerve head (Figure 2C, asterisk) and the fovea (Figure 2E, arrow). SDOCT B-scans of the affected donor revealed that retinal vessels appear to be absent or atrophic in the affected donor retina even around the optic nerve (Figure 2D, asterisk). The retina of the affected donor also displayed absence of a photoreceptor layer, and degeneration of the choroid in the macular area (Figure 2F, arrow) when compared to the control eye (Figure 2E, arrow).
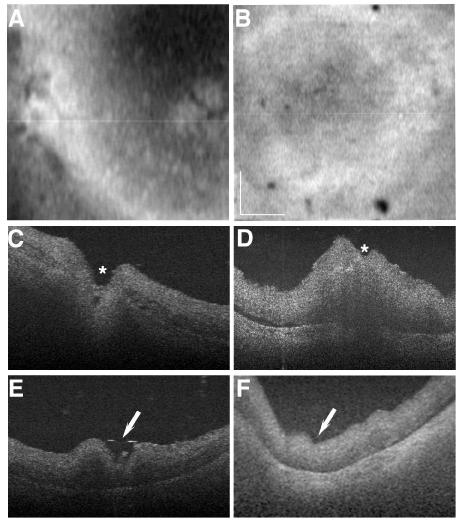
Figure 2: Retinal disorganization of the affected donor retina. Ex-vivo SDOCT
en face imaging of the retina of affected donor (B) and age-matched
control (A) control eye. C-F: SD-OCT B-scans of affected donor (D, F) and
age-matched control (C, E) eye. Asterisks show optic nerve head, and white
arrows show macula. Bar = 0.5 cm.
Gross pathology of resin-embedded donor tissue was analyzed and compared to the age-matched control (Figure 3). The retina of the control eye displayed each of the usual retinal lamina and the RPE in the perifovea and periphery (Figure 3A and C). The retina of the affected donor displayed a highly degenerated retina with disorganization of the retinal laminae and gliosis in both the perifovea and periphery (Figure 3B and D). Extensive degeneration could be observed in the perifovea of the affected donor. However, a few photoreceptor nuclei were still detectable. The inner and outer retinal layers of the affected donor were reduced but still present while the photoreceptors were absent. A thin, continuous area of pigmented RPE cells was observed in the perifovea but not in some areas of the periphery.
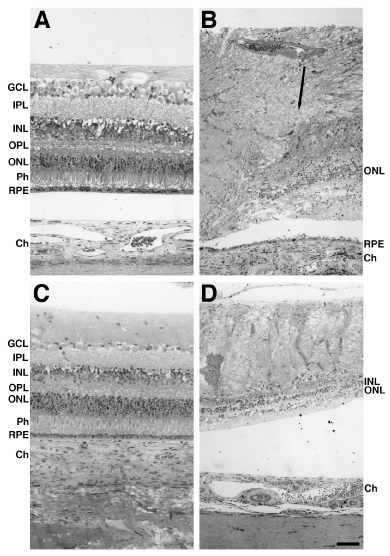
Figure 3: Morphological abnormalities in the retinas of affected donor.
Toluidine blue staining of 1μm plastic sections of age-matched control retina
in the perifovea (A) and periphery (C) compared to affected donor retina in
the perifovea (B) and periphery (D) were analyzed by bright-field microscopy.
Black arrow delineates the edge of the macular region.
GCL: Ganglion Cell Layer; IPL: Inner Plexiform Layer; INL: Inner Nuclear
Layer; OPL: Outer Plexiform Layer; ONL: Outer Nuclear Layer; Ph:
Photoreceptors; RPE: Retinal Pigment Epithelium. Bars =100 μm.
Next, we carried out immunohistological analysis of the retinas of both the control and affected donor. With respect to the control eye, cone arrestin was distributed along the entire plasma membrane of this cell, from the tip of the outer segment to the synaptic base both in the perifovea and periphery (Figure 4A and C). Cones were present in the perifovea of the affected donor; however, they were sparse and had lost their vertical orientation (Figure 4B, arrows). Additionally, the synapses could not be visualized. Cones were mostly absent from the periphery of the affected donor retina, and the few cones observed displayed stubby morphology (Figure 4D, arrows).
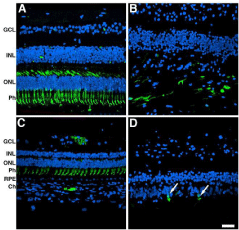
Figure 4: Significant alterations in the cones both in the perifovea and in the
periphery of affected donor. Representative immunofluorescent staining of
cone arrestin in control retina in the perifovea (A) and periphery (C) compared
to affected donor retina in the perifovea (B) and periphery (D) were analyzed
by confocal microscopy.
GCL: Ganglion Cell Layer; INL: Inner Nuclear Layer; ONL: Outer Nuclear
Layer; Ph: Photoreceptors; RPE: Retinal Pigment Epithelium; Ch: Choroid.
Bar = 40 μ.
The rod outer segments were visualized through labeling with rhodopsin antibodies (Figure 5). Control retinas displayed restricted distribution of rhodopsin to the outer segment of rods both in the perifovea and periphery (Figure 5A and C). Although rods were mostly absent from both the perifovea and the periphery of the affected donor (Figure 5B and D), a few isolated labeled cells remained in the periphery (Figure 5D, arrow).
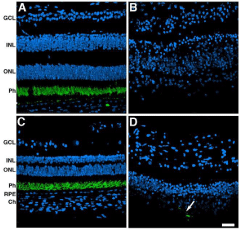
Figure 5: Significant decrease in rods both in the perifovea and in the
periphery of affected donor. Representative immunofluorescent staining of
rhodopsin in control retina in the perifovea (A) and periphery (C) compared to
affected donor retina in the perifovea (B) and periphery (D) were analyzed by
confocal microscopy. White arrow points to isolated rhodopsin-labeled cell.
GCL: Ganglion Cell Layer; INL: Inner Nuclear Layer; ONL: Outer Nuclear
Layer; Ph: Photoreceptors; RPE: Retinal Pigment Epithelium; Ch: Choroid.
Bar = 40 μm.
Labeling with calbindin D-28k (green) and GFAP (red) antibodies was done to visualize second order neurons and Müller cells, respectively (Figure 6). Control retinas labeled with calbindin D-28k antibody displayed localization to sparse neurons in the ganglion cell layer, to amacrine, bipolar and cones in the perifovea (Figure 6A). In the affected donor, less calbindin D-28k positive cells were observed in the ganglion cell layer, amacrine, and bipolar cells (Figure 6B). However, atrophic, round calbindin D-28k-labeled structures were present in the inner plexiform layer (Figure 6B, arrowheads). This labeling was also sparsely present in peripheral neurons of the control retinal ganglion cell layer, in amacrine and bipolar cells (Figure 6C). In the periphery of the affected donor, calbindin D-28k positive cells were decreased in the ganglion cell layer, amacrine and bipolar cells. However, calbindin D-28k-labeled structures were present in the inner plexiform layer (Figure 6D, arrowheads). The Müller cells had undergone reactive gliosis throughout the affected donor retina, and their hypertrophied processes were strongly positive for GFAP in both the perifovea and periphery.

Figure 6: Altered distribution in second order neurons and increased
presence of Müller cells, which have undergone reactive gliosis in the eye
of affected donor. Representative immunofluorescent staining of calbindin
D-28k (green) and GFAP (red) in control retina in the perifovea (A) and
periphery (C) compared to affected donor retina in the perifovea (B) and
periphery (D) were analyzed by confocal microscopy. White arrowheads point
to calbindin D-28k-labeled structures present in the inner plexiform layer.
GCL: Ganglion Cell Layer; IPL: Inner Plexiform Layer; INL: Inner Nuclear
Layer; ONL: Outer Nuclear Layer; Ph: Photoreceptors; RPE: Retinal Pigment
Epithelium; Ch: Choroid. Bar = 40 μm.
The RPE was visualized through labeling with antibodies to RPE65 (Figure 7). Control retinas displayed a continuous layer of strong antibody binding in the RPE of both the perifovea and periphery (Figure 7A and C). The affected donor retina also demonstrated the presence of RPE65 in the RPE of both the perifovea (Figure 7B) and periphery regions (Figure 7D). In the affected donor, several layers of RPE65 positive cells were observed on top of the RPE cells of both perifovea and periphery (Figure 7B and D, arrow). Besides, some RPE65-labeled cells were also present in the retina (Figure 7B and D, arrowheads). Further analysis of the RPE compared the relative amount of autofluorescence in both the postmortem control eye with the RPE cells from the affected donor both in the macula (Figure 8A and B) and the periphery (Figure 8C and D). A few autofluorescent granules or lipofuscin were visible in the cytoplasm of the control RPE in the macula (Figure 8A, arrows). Although still present, in the periphery the control RPE displayed even fewer of these autofluorescent granules (Figure 8C, arrows). However, RPE of the affected donor showed a paucity of autofluorescence in both the macula (Figure 8B) and in the periphery (Figure 8D).
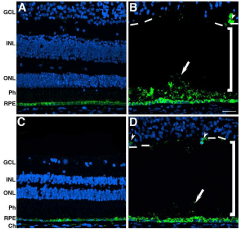
Figure 7: Presence of RPE cells and cells expressing RPE65 in the retina
of the affected donor. Representative immunofluorescent staining of RPE65
in control retina in the perifovea (A) and periphery (C) compared to affected
donor retina in the perifovea (B) and periphery (D) were analyzed by confocal
microscopy. White arrows show RPE65 positive cells on top of the RPE,
white arrowheads show RPE65 positive cells in the retina, brackets delineate
the area between the detached retina and the RPE, and hashed white lines
indicate the edges of the detached retina.
GCL: Ganglion Cell Layer; INL: Inner Nuclear Layer; ONL: Outer Nuclear
Layer; Ph: Photoreceptors; RPE: Retinal Pigment Epithelium; Ch: Choroid.
Bar = 40 μ.
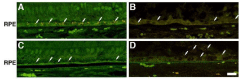
Figure 8: Decreased presence of autofluorescent granules in the RPE in the
eyes of the affected donor. Representative fluorescence in the green channel
(excitation 495 nm/emission 680 nm) of cryosections of a control retina in
the perifovea (A) and periphery (C) compared to affected donor retina in the
perifovea (B) and periphery (D) were analyzed by confocal microscopy. White
arrows show autofluorescent granules in the cytoplasm of retina. Bar = 20
μm.
Labeling with centrin-2 (green) and cone arrestin (red) antibodies was done to visualize the connecting cilium of the photoreceptor cells and cones, respectively (Figure 9). Control retinas labeled with centrin-2 antibody displayed localization to the connecting cilium of the photoreceptor cells of both the perifovea (Figure 9A) and periphery (data not shown). Cone cell bodies were labeled with the cone arrestin antibody as described above. However, the affected donor retina did not display visible labeling for centrin-2 (Figure 9B). High magnification observation of this labeling revealed the continuous presence of centrin-2 along the entire connecting cilium in both perifovea and periphery of control retinas (Figure 9C and E). In contrast, the affected donor retina displayed the dispersed localization of a few vesicles throughout the cell body of some of the few cone cells still present in both the perifovea (Figure 9D, arrows) and periphery (Figure 9F, arrows).

Figure 9: Absence of photoreceptor cells expressing centrin-2 in the retina of
the affected donor. Representative immunofluorescent staining of centrin-2
(green) and cone arrestin (red) in control retina in the perifovea (A, C) and
periphery (E) compared to affected donor retina in the perifovea (B, D) and
periphery (F) were analyzed by confocal microscopy. White arrowheads
show centrin-2 positive vesicles in the cone photoreceptors cell body labeled
by cone arrestin.
ONL: Outer Nuclear Layer; CC: Connecting Cilium; POS: Photoreceptor
Outer Segments. Bar (A, B) = 10 μm; (C- F) = 5 μm.
Discussion
Here, we report the clinical, morphological, histological and immunohistological findings of a 3 year-old affected eye donor. He was found to have profound visual impairment and was diagnosed with LCA at the age of 1.5 years. LCA is a congenital retinal dystrophy characterized by nystagmus, photophobia, loss of vision and profoundly reduced electroretinographic responses. To date, 23 genes have been identified to cause LCA. The majority of cases of LCA are inherited in an autosomal recessive manner; however, a few dominant cases have been reported [3-6]. Retinal dystrophies, including LCA, can be isolated or part of a syndrome. Given the additional medical history of this donor, he most likely was affected with one of the syndromic retinal dystrophies, more specifically Joubert syndrome in view of the cerebellar atrophy and vermis hypoplasia.
Joubert syndrome is an autosomal recessive condition, which exhibits both genetic and phenotypic heterogeneity. It is characterized by a “molar tooth” sign seen on brain MRI, which is a result of cerebellar vermis hypoplasia, thick and maloriented superior cerebellar peduncles, and an abnormally deep interpeduncular fossa [7]. Individuals with Joubert syndrome may have hypotonia in infancy, ataxia, developmental delay, microcephaly, retinal dystrophy, multicystic kidney disease, polydactyly and episodic hyperpnoea with hyperventilation [8]. To date, 26 genes have been identified to be associated with Joubert or Joubert-related syndrome, all of which encode for proteins of the primary cilium [9]. This observation links these disorders to the field of ciliopathies [10]. Primary cilia are highly-conserved organelles made up of a core composed of a microtubule-based axoneme emerging from a basal body derived from the mother centriole and ensheathed by the ciliary membrane [11]. The proteins encoded by all the Joubert syndrome genes have been shown to either localize to the primary cilium and basal body or directly interact with components of the primary cilium and basal body [12]. In addition, it has been reported that mutations in the mouse orthologs of two Joubert syndrome genes, namely rpgrip1l and arl13b, disrupt cilium number or morphology. The retina of the affected donor analyzed here displayed significant decrease in the photoreceptor cells together with lack of cilium in the few remaining photoreceptors. This observation is indicative that this donor had a ciliopathy, and most likely Joubert syndrome. In addition, much of the clinical history of this donor—such as in view of the other clinical findings such as hypotonia, developmental delay, respiratory distress, and the cerebellar abnormalities noted on findings on brain MRI— also suggested that he most likely had Joubert syndrome.
A review of the medical literature does not provide comprehensive and significant data regarding the retinal dystrophy in patients with Joubert syndrome. Most of what is reported is that patients with Joubert have a retinal dystrophy similar to LCA [13]. However, a recent study carried out in fibroblasts and tissues isolated from a patient carrying a homozygous splice-site mutation in PDE6D failed to identify changes in both the number and gross morphology of primary cilia [14]. Because of the wide genetic and clinical heterogeneity of this clinical syndrome, it is unclear if patients with Joubert have a characteristic fundus appearance or other pathognomonic features seen on retinal imaging. Here, the morphological analysis of the affected donor’s eyes revealed an atrophic maculopathy associated with significant decrease in photoreceptor cells. There was disorganization of the retina histologically and a significant decrease of rods and cones, with few surviving cones in the perifovea region but close to complete absence of rods in the retina. The RPE layer displayed thinning in some regions of the periphery and decreased pigmentation in most areas. RPE65 labeled cells were detected in the RPE layer and in cells present in the retina. We observed the presence of small amounts of lipofuscin in the control RPE. However, RPE of the affected donor showed a paucity of lipofuscin granules. Lipofuscin represents lysosomal residual bodies related to photoreceptor phagocytosis and digestion. Old eyes are characterized by high concentration of lipofuscin granules [15-17]. Finally, centrin-2 labeling was mostly absent in this affected donor.
Conclusion
We have provided the clinical, histopathologic and immunocytochemical findings in donor eyes from a boy with a ciliopathy characterized by multiple congenital abnormalities, including a retinal dystrophy. While we could not document a molecular diagnosis, the clinical history and clinical findings are highly suggestive of Joubert syndrome.
Acknowledgment
The authors thank Y. Li for excellent technical support. This work was supported by the Foundation Fighting Blindness, Research to Prevent Blindness, Wolf Family Foundation and The National Eye Institute.
References
- Chan S, Freund PR, MacDonald I. Advances in the genetics of eye diseases. Curr Opin Pediatr. 2013; 25: 645-652.
- Bonilha VL, Rayborn ME, Bell BA, Marino MJ, Fishman GA, Hollyfield JG. Retinal Histopathology in Eyes from a Patient with Stargardt disease caused by Compound Heterozygous ABCA4 Mutations. Ophthalmic Genet. 2015.
- Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998; 63: 1307-1315.
- Tzekov RT, Liu Y, Sohocki MM, Zack DJ, Daiger SP, Heckenlively JR, et al. Autosomal dominant retinal degeneration and bone loss in patients with a 12-bp deletion in the CRX gene. Invest Ophthalmol Vis Sci. 2001; 42: 1319-1327.
- Nichols LL 2nd, Alur RP, Boobalan E, Sergeev YV, Caruso RC, Stone EM, et al. Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Hum Mutat. 2010; 31: E1472-1483.
- Zou X, Yao F, Liang X, Xu F, Li H, Sui R, et al. De novo mutations in the cone-rod homeobox gene associated with leber congenital amaurosis in Chinese patients. Ophthalmic Genet. 2015; 36: 21-26.
- McGraw P. The molar tooth sign. Radiology. 2003; 229: 671-672.
- Rehman Iu, Bett Z, Husen Y, Akhtar AS, Khan FA. The 'molar tooth sign' in Joubert syndrome. J Pak Med Assoc. 2009; 59: 851-853.
- Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis. 2014; 9: 72.
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008; 51: 1-23.
- Fry AM, Leaper MJ, Bayliss R. The primary cilium: guardian of organ development and homeostasis. Organogenesis. 2014; 10: 62-68.
- Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009; 16: 143-154.
- Valente EM, Dallapiccola B, Bertini E. Joubert syndrome and related disorders. Handb Clin Neurol. 2013; 113: 1879-1888.
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014; 35: 137-146.
- Wing GL, Blanchard GC, Weiter JJ. The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1978; 17: 601-607.
- Weiter JJ, Delori FC, Wing GL, Fitch KA. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest Ophthalmol Vis Sci. 1986; 27: 145-152.
- Bonilha VL, Rayborn ME, Li Y, Grossman GH, Berson EL, Hollyfield JG. Histopathology and functional correlations in a patient with a mutation in RPE65, the gene for retinol isomerase. Invest Ophthalmol Vis Sci. 2011; 52: 8381-8392.