
Research Article
Austin J Clin Pathol. 2014;1(3): 1012.
Study on the Use of Cellular Respiration as a Surrogate Biomarker in Drug Development
Al-Hammadi S1, Alfazari AS2, Shaban S3 and Souid A-K1*
1Department of Pediatrics, UAE University, UAE
2Department of Medicine, UAE University, UAE
3Department of Medical Education, UAE University, UAE
*Corresponding author: Souid A-K, Department of Pediatrics, UAE University, Tawam campus, Al-Ain, Abu Dhabi, UAE
Received: May 25, 2014; Accepted: June 30, 2014; Published: July 02, 2014;
Abstract
Cellular respiration (mitochondrial O2 consumption or oxidative phosphorylation) is the process of delivering nutrients and O2 to the mitochondria, oxidation of reduced metabolic fuels, passage of electrons to O2, and synthesis of ATP. These vital processes can serve as surrogate biomarkers for identifying cellular responses to drugs. This study investigated divergent compounds to learn whether their modes-of-action or adverse events could be linked to altered cellular bioenergetics. Small tissue fragments were collected from various organs of C57BL/6 mice in RPMI with and without designated drugs. Cellular respiration was then measured with the aid of a phosphorescence O2 analyzer, using Pd (II) complex of meso-tetra-(4-sulfonatophenyl)-tetrabenzoporphyrin as an O2 probe. The phosphoinositide 3-kinase/mammalian target of rapamycin (Pi3K/mTOR) inhibitor GSK2126458 (50nM) repressed heart cellular respiration (p<0.001). This finding is consistent with the role of Pi3K/PTEN/Akt/mTOR pathway in cellular metabolism including insulin-dependent glucose uptake. The β2-adrenergic receptor agonist salbutamol (29 μM) and the bronchodilator magnesium sulphate (20mM) both inhibited lung cellular respiration (p≤0.006). The N-methyl-D-aspartate receptor antagonist ketamine (210 to 1,050 μM) had no significant effect on forebrain cellular respiration (p≤0.132).Although valproate induced histologic liver abnormalities, a therapeutic concentration of the drug (600 μM) had no effect on hepatocyte respiration (p=0.916). Similarly, the hepatotoxic drug azidothymidine (a nucleoside reverse transcriptase inhibitor; 100 μM) had no effect on hepatocyte respiration (p=0.297). Thus, cellular respiration can sense the activity and toxicity of some drugs. This analytical tool needs to be incorporated in drug development as a pharmacodynamic measure in targeted and off-targeted tissues.
Keywords: PI3K/mTOR Inhibitors; Salbutamol; MgSO4; Ketamine; Valproate; Azidothymidine
Abbreviations
Pi3K: Phosphoinositide 3-kinase; mTOR: Mammalian Target of Rapamycin; NMDA: N-methyl-D-aspartate; ATP: Adenosine 5'-triphoshate; NRTI: Nucleoside Reverse Transcriptase Inhibito
Introduction
Studies addressing the effects of drugs on cellular bioenergetics [the metabolic reactions involved in energy conversion or transformation including cellular respiration and accompanying adenosine 5'-triphoshate (ATP) synthesis] are limited [1-2]. These vital processes are involved in many drug mechanisms and adverse events. Therefore, it is reasonable to incorporate measurements of cellular energy into drug development programs. This study investigated the effects of several drugs on cellular respiration. Its main purpose was to use mitochondrial O2 consumption as a surrogate biomarker for assessing drug activity and toxicity.
Cellular respiration has been previously measured in specimens from the heart, liver, and kidney[3-4]. Therefore, drugs that target these organs are potential candidates for testing. In contrast, the hematopoietic system and pancreas are not readily permissible organs a brief account of these compounds is given below.
GSK2126458 potently inhibits phosphoinositide 3-kinase (Pi3K) and mammalian target of rapamycin (mTOR) [5]. This novel class of drugs is in clinical trials for treatment of various human diseases, as monotherapy or in combination with other cytotoxic agents. This therapeutic approach targets critical processes, such as survival pathways (e.g., Pi3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK) and cellular bioenergetics. For example, engagement of insulin with its receptor activates Pi3K, allowing cellular glucose uptake and other insulin signaling. Consistently, the activities of Pi3K inhibitors are expected to include impaired cellular bioenergetics [6-8]. GSK2126458 was studied here using heart cellular respiration since this organ consumes high metabolic energy and this type of drugs is known to have cardiac toxicity [9-10].
Salbutamol sulphate (a selective β2-adrenergic receptor agonist) and magnesium sulphate (Epsom salt) are routinely used medications for treatment of bronchospasm [11]. Plasma salbutamol concentration following repetitive dosing is about 0.2 μM [12]; the drug however, is commonly given by repetitive inhalations, which result in higher lung tissue concentrations. Therapeutic serum magnesium sulphate concentrations range from 2.0 to 3.5mM; higher dosing is associated with loss of deep tendon reflexes and cardiorespiratory arrest [13]. To our knowledge, the effects of these drugs on lung tissue cellular respiration have not been adequately previously studied.
Adverse events of the antiepileptic drug valproate include hepatotoxicity, mitochondrial dysfunction and complex metabolic derangements [14-15]. Its direct toxicity has been demonstrated in isolated liver mitochondria [16]. In cultured fibroblasts, the drug increases mitochondrial biogenesis [17]. Its effect on cellular respiration in isolated liver tissue is unknown. In one study, peak plasma valproate levels ranged from 465 μM to 1,118 μM [18].
Similarly, hepatic failure associated with the nucleoside reverse transcriptase inhibitors (NRTI), such as azidothymidine has been attributed to mitochondrial toxicity, which includes inhibition of mitochondrial DNA polymerases [19-20]. In one study, the plasma azidothymidine level was about 8 μM [21]. Valproate and azidothymidine are studied here using liver cellular respiration; (they are tested alone and in combination since both are potentially hepatotoxic). The N-methyl-D-aspartate receptor antagonist (NMDA) ketamine is studied using forebrain cellular respiration [22], as previously described [23].
Methods
Reagents and solutions
The Pi3K/mTOR inhibitor GSK2126458 (m.w. 505.5; cat. #HY- 10297) was purchased from MedChem Express, LLC (Princeton, NJ); the compound was dissolved in DMSO at 5 mg/mL (9.9mM) and stored at -20°C. Salbutamol BP (as sulphate, m.w. 576.7) 0.5% w/v (8.7mM) was purchased from Glaxo Operations UK Limited (Barnard, UK); the drug was stored at 25oC. Magnesium Sulphate Injection (as pentahydrate, m.w. 246.5) 50% w/v (2.0 M) was purchased from Martindale Pharmaceuticals (Romford, UK); the drug was stored at 25oC. Ketamine HCl Injection (m.w. 274.2) containing 50 mg ketamine base (m.w. 237.7) per mL (210mM) was purchased from JHP Pharmaceuticals (Rochester, MI, US). Sodium valproate (400 mg powder and solvent for solution for injection vials; m.w. 166.2) was purchased from Sanofi (Surrey, UK); the drug was dissolved in Water for Injection immediately before use and the powder was stored at 25oC in air-sealed vial. The antiretroviral drug azidothymidine (AZT; m.w. 267.2; 10 mg/mL of Water for Injection; 37.4mM) was purchased from Glaxo Operations UK Limited (Barnard, UK).
Pd (II) complex of meso-tetra- (4-sulfonatophenyl) -tetrabenzoporphyrin (Pd phosphor) was purchased from Porphyrin Products (Logan, UT). Pd phosphor (2.5 mg/mL = 2mM), NaCN (1.0 M) and glucose oxidase (10 mg/mL) solutions were prepared as previously described and stored at -20°C [3-4]. RPMI 1640 medium and remaining reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Mice
C57BL/6 (9-10 weeks old) mice were housed at the animal facility in rooms maintained at 22°C, 60% humidity and 12-h light-dark cycles. The mice had ad libitum access to standard rodent chow and filtered water. The study received approval from the Animal Ethics Committee - United Arab Emirates University - College of Medicine and Health Sciences.
Tissue collection and processing
Urethane (25% w/v, 100 μL per 10 g) was used to anesthetize the mice. Tissue fragments (about 10 to 25 mg each) were then cut manually with sterile scalpels (Swann-Morton, Sheffield, England) and immediately processed for measuring cellular respiration in the presence and absence of designated concentrations of the studied drugs. Alternately, liver specimens were incubated at 37°C in 50mL RPMI (continuously gassed with 95% O2: 5% CO2) with and without the drugs for up to 6 h. At designated times; samples were removed from the incubation solution and processed for measuring cellular respiration. Some samples were also processed for histology (hematoxylin & eosin staining), cellular ATP and cellular GSH as previously described [3-4].
Cellular respiration
Phosphorescence O2 analyzer was used to measure cellular mitochondrial O2 consumption as previously described [3-4]. Briefly, samples were exposed to 600/min flashes. O2 detection was with the aid of Pd phosphor (absorption 625 nm; emission maximum 800 nm). The phosphorescence was detected by Hamamatsu photomultiplier tube. The phosphorescence decay rate (1/τ) was single exponential; 1/τ was linear with dissolved O2: 1/τ = 1/τ° + kq[O2], 1/τ = phosphorescence decay rate in presence of O2, 1/τ° = phosphorescence decay rate in absence of O2, and kq = second-order O2 quenching rate constant (s-1 μM-1) [24]. A Microsoft Visual Basic 6 program was developed with the aid of Microsoft Access Database 2007 and Universal Library components (Universal Library for Measurements Computing Devices). These tools allowed direct reading from the PCI-DAS 4020/12 I/O Board (PCI-DAS 4020/12 I/O Board) [25]. O2 measurements were performed at 37°C in glass vials sealed from air. Respiratory substrates were endogenous metabolic fuels and the glucose present in RPMI. [O2] decreased linearly with time; this zero-order process was inhibited by cyanide (CN), confirming O2 consumption occurred in the mitochondrial respiratory chain. The rate of respiration (k; μM O2 min-1) was the negative of the slope d [O2]/dt. The value of k was divided by specimen weights, giving kc (μM O2 min-1 mg-1). The data were analyzed on SPSS statistical package (version 19), using the nonparametric (two independent samples) Mann-Whitney test.
Results
Figure 1A shows representative runs of heart muscle cellular mitochondrial O2 consumption with and without the Pi3K/mTOR inhibitor GSK2126458. Each run represented a specimen that was collected from a C57BL/6 mouse and immediately processed for measuring cellular respiration in the presence and absence of 50nM GSK2126458. A summary of the results is shown in Figure 1B and Table 1. The rate of respiration (kc; μM O2 min-1 mg-1; mean ± SD) without addition was 0.19 ± 0.04 (n = 14 mice) and with the addition of GSK2126458 was 0.08 ± 0.03 (n = 17 mice; p<0.001). Thus, GSK2126458 decreased heart cellular respiration by about 58%.
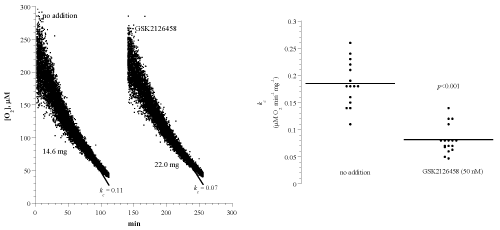
Figure 1: Effects of the Pi3K/mTOR inhibitor GSK2126458 on heart muscle cellular respiration of C57BL/6 mouse. Panel A: Each run represented a fragment that was collected from the apex of the heart and placed in RPMI with or without 50 nM GSK2126458. Cellular respiration was measured at 37°C immediately after each specimen collection. Rate of respiration (k; μM O2 min-1) was the negative of the slope of [O2] vs. t. The values of kc (μM O2 min-1 mg-1) and the weight of each sample are shown at the bottom of each run. Panel B: Summary of all measurements; lines are means.
![]()
Tissue
Incubation
time (h)
Drug Concentration
kc
(�M O2 min-1 mg-1)
Inhibition
(%)
p-value
GSK2126458
(Pi3K/mTOR inhibitor)
Heart
2.3 - 7.7
0
0.19 ± 0.04 (14)
-
-
50 nM
0.08 ± 0.03 (17)
58
<0.001
Salbutamol
(�2-adrenergic receptor agonist)
Lung
=1.0
0
0.18 ± 0.04 (4)
-
-
29 �M
0.08 ± 0.03 (7)
56
0.006
Magnesium sulphate
(a bronchodilator)
Lung
=1.0
0
0.20 ± 0.04 (7)
-
-
20 mM
0.12 ± 0.03 (7)
40
0.001
Ketamine
(N-methyl-D-aspartate receptor antagonist)
Brain
<1.0
0
0.46 ± 0.08 (6)
-
-
210-1,050 �M
0.36 ± 0.12 (6)
22
0.132
Valproate
(an anticonvulsant)
Liver
=6.0
0
0.19 ± 0.08 (44)
-
-
600 �M
0.19 ± 0.06 (20)
0
0.916
Azidothymidine
(a nucleoside analog reverse-transcriptase inhibitor)
Liver
=6.0
0
0.19 ± 0.08 (44)
-
-
100 �M
0.18 ± 0.07 (36)
5
0.297
Table 1: Effects of studied drugs on cellular respiration of their targeted tissue. Tissue fragments (from C57BL/6 mice) were collected in RPMI and processed for measuring cellular respiration with and without the designated drugs. The values of kc are mean ± SD (n).
Figure 2A shows representative runs of lung tissue cellular mitochondrial O2 consumption with and without salbutamol. The experimental conditions were as discussed above. A summary of the results is shown in Figure 2B and Table 1. The rate of respiration (kc; μM O2 min-1 mg-1) without addition was 0.18 ± 0.04 (n = 4 mice) and with the addition of 29 μM salbutamol was 0.08 ± 0.03 (n = 7 mice; p=0.006). Thus, salbutamol decreased lung cellular respiration by about 56%.
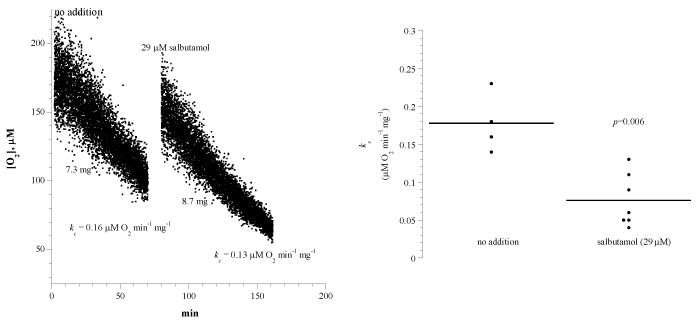
Figure 2: Effects of the β2-adrenergic receptor agonist salbutamol on lung cellular respiration. Panel A: Each run represented a fragment that was collected from the lung and placed in RPMI with or without 21 μM salbutamol. Cellular respiration was measured at 37°C immediately after each specimen collection. Panel B: Summary of all measurements. Please see Legend to Figure 1.
Figure 3A shows representative runs of lung cellular mitochondrial O2 consumption with and without magnesium sulphate (20mM). The experimental conditions were as discussed above. The addition of cyanide halted cellular respiration, confirming the oxidation occurred in the mitochondrial respiratory chain. The addition of glucose oxidase (catalyzes the reaction D-glucose + O2 → D-glucono-δ-lactone + H2O2) depleted remaining O2 in the solution. A summary of the results is shown in Figure 3B and Table 1. The rate of respiration (kc; μM O2 min-1 mg-1) without addition was 0.20 ± 0.04 (n = 8 mice) and with the addition of magnesium sulphate was 0.11 ± 0.03 (n = 8 mice; p=0.001). Thus, the drug decreased lung cellular respiration by about 45%.
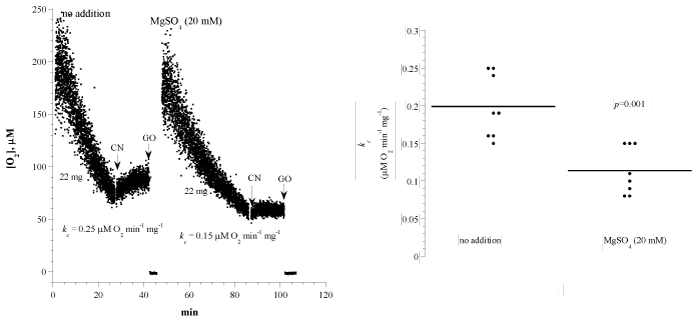
Figure 3: Effects of the bronchodilator magnesium sulphate on lung cellular respiration. Panel A: Each run represented a fragment that was collected from the lung and placed in RPMI with or without 42 μM MgSO4. Cellular respiration was measured at 37°C immediately after each specimen collection. Panel B: Summary of all measurements. Please see Legend to Figure 1.
Figure 4A shows runs of forebrain tissue cellular mitochondrial O2 consumption with and without ketamine (210 to 1,050 μM). Each run represented a specimen that was collected from a C57BL/6 mouse and immediately processed for measuring cellular respiration. Ketamine was injected directly into the vial during the O2 run. The rate of respiration (kc; μM O2 min-1 mg-1) before ketamine injections was 0.46 ± 0.08 and after ketamine injections was 0.36 ± 0.12 (n = 6 mice; p=0.132). Thus, ketamine had no effect on forebrain cellular respiration.
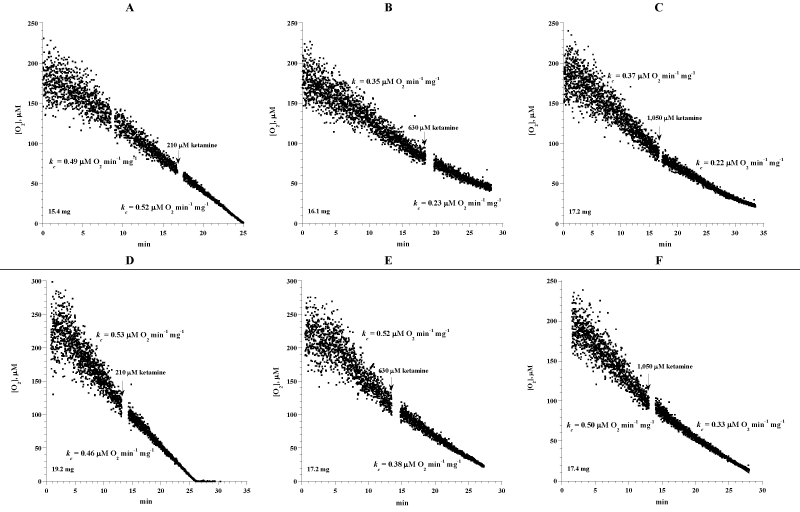
Figure 4: Effects of N-methyl-D-aspartate receptor antagonist ketamine on forebrain cellular respiration. Each run represented a fragment that was collected from the forebrain and placed in RPMI. Cellular respiration was measured at 37oC immediately after each specimen collection. Designated concentrations of ketamine were added during the runs. The values of kc (μM O2 min-1 mg-1) before and after ketamine injection and the weight of each sample are shown at the bottom of each run.
In an in vivo experiment, four mice were injected intraperitoneally with ketamine as previously described [23]. Forebrain specimens were then collected at 30, 60, 90, and 120 min post injection and processed immediately for measuring cellular respiration. The rate of respiration (kc; μM O2 min-1 mg-1) at 30 min was 0.50 (dose = 0.76 μmol/g), at 60 min was 0.57 (dose = 0.76 μmol/g), at 90 min was 0.44 (dose = 0.69 μmol/g), and at 120 min was 0.34 (dose = 0.74 μmol/g). The corresponding value for untreated mouse was 0.38 μM O2 min-1 mg-1. In another in vivo experiment, both cellular respiration and ATP was determined. At 30 min, the value of kc was 0.55 μM O2 min-1 mg-1 and ATP was 121 pmol mg-1 (dose = 1.7 μmol/g). At 60 min, the value of kc was 0.58 μM O2 min-1 mg-1 and ATP was 177 pmol mg-1 (dose = 0.9 μmol/g). At 90 min, the value of kc was 0.45 μM O2 min-1 mg-1 and ATP was 251 pmol mg-1 (dose = 1.9 μmol/g). At 120 min, the value of kc was 0.34 μM O2 min-1 mg-1 and ATP was 160 pmol mg-1 (dose = 1.2 μmol/g). The corresponding kc and ATP values in untreated mouse were 0.38 μM O2 min-1 mg-1 and 214pmol mg-1, respectively. It is worth noting that the usual anesthetic dose in mice is about 0.85μmol/g [22]. Thus, the in vivo anesthetic dosing of ketamine also had no effects on forebrain cellular respiration. In our hand, dosing >2 μmol/kg was lethal in <10 min.
Figure 5A shows representative runs of liver cellular mitochondrial O2 consumption in samples incubated in vitro with and without azidothymidine (100 μM, AZT), valproate (600 μM, Val) or combination of both drugs. In this experiment, liver specimens were collected from one mouse and incubated at 37°C in RPMI with and without the designated compounds. At the designated times, specimens were removed from the incubation solutions, rinsed with RPMI and processed for measuring O2 consumption at 37°C. A summary of all experiments is shown in Figure 5B (15 separate experiments involving 15 mice) and Table 1. The rate of respiration (kc; μM O2 min-1 mg-1) without addition was 0.19 ± 0.08 (n = 44 runs), with azidothymidine was 0.18 ± 0.07 (n = 36 runs; p=0.297), with valproate was 0.19 ± 0.06 (n = 20 runs; p=0.916), and with both compounds was 0.18 ± 0.05 (n = 20 runs; p=0.548). Thus, liver cellular respiration was not affected by azidothymidine and valproate.
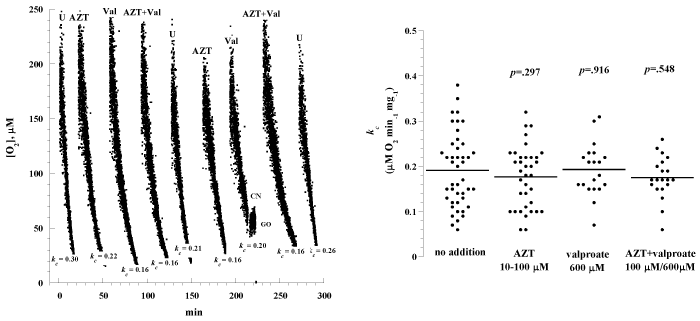
Figure 5: Effects of the hepatotoxic drugs valproate and azidothymidine on liver cellular respiration. Panel A: Representative runs of liver cellular respiration. Multiple liver specimens were collected from a C57BL/6 mouse and incubated at 37°C in RMPI with and without 100 μM azidothymidine (AZT), 600 �M valproate (Val), or combination of both drugs. At designated time periods, the specimens were removed from the incubation solution, rinsed with RMPI and placed in the O2 vial for measuring cellular respiration. The values of kc (μM O2 min-1 mg-1) are shown at the bottom of each run. Panel B: Summary of all measurements; the lines are means.
In another experiment, liver specimens were incubated at 37°C in RPMI with and without 600 μM valproate for 4 h. At the end of the incubation period, the samples were processed for histology and determination of intracellular GSH and caspase activity. Compared to untreated, the treated fragment revealed more prominent cellular disintegration, spottier cell necrosis, and steatosis (Figure 6). Hepatocyte GSH in untreated sample was 672pmol mg-1 and in treated sample was 640pmol mg-1. Hepatocyte GSH at 0 h (immediately after tissue collection) was 1,176pmol mg-1. Intracellular caspase activity was also similar in treated and untreated samples (data not shown). Thus, the valproate-induced morphologic abnormalities were not associated with measurable changes in cellular respiration, GSH concentration, and caspase activity.
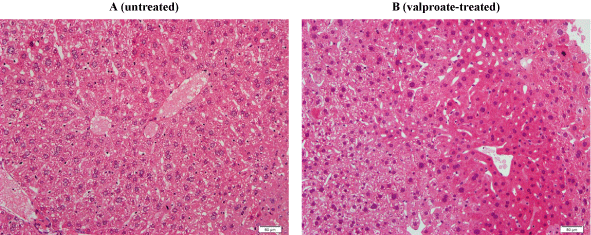
Figure 6: Liver histology with and without valproate. H&E staining sections of liver fragments incubated at 37°C with (B) and without (A) 600 μM valproate for 4 h. Panel A: Untreated fragment showing preserved hepatic architecture with noticeable cytoplasmic ballooning, vacuolar degeneration, and spotty cell necrosis. The cell membrane is intact and the nuclear details are preserved. These findings reflected a slight deterioration in histology compared to 0 h (not shown). Panel B: Treated fragment showing preserved hepatic architecture with cellular disintegration, spottier cell necrosis, and steatosis. Histology of treated specimen was worse than untreated specimen at 4 h.
Discussion
The mitochondria use energy derived from oxidations in the respiratory chain to generate ATP (oxidative phosphorylation). These vital organelles also sense injury signals and release pro-apoptotic molecules that trigger the caspase (cysteine-dependent aspartate-directed protease) cascade. Caspase activation promotes mitochondrial impairments [19-20].
Many adverse events of drugs are attributed to mitochondrial toxicity [1]. This fact supports the need for reliable methodologies that assess early effects of compounds on cellular bioenergetics [2]. This aim is addressed here by using mitochondrial O2 consumption as a surrogate biomarker for measuring cellular responses to various drugs. The results of all studied compounds are summarized in Table 1.
The rapidly emerging small molecule inhibitors of protein kinases (e.g., the Pi3K/mTOR inhibitor GSK2126458) are active in a variety of human diseases including cancer and inappropriate immune responses. The therapeutic gains of these agents have been linked to suppressing cellular metabolism, preventing cell growth and inducing apoptosis [5-8]. The results here show GSK2126458 invokes potent inhibitory effects on heart muscle cellular mitochondrial O2 consumption (Figure 1). This result is consistent with the known role of Pi3K/AKT pathway in regulating cardiomyocyte death [9-10]. The high concentrations of salbutamol (29 μM) and magnesium sulphate (20mM) used here inhibited lung cellular respiration (Table 1). It is unclear, however, if these effects are relevant to human therapeutic dosing. High doses of ketamine, on the other hand, had no effect on forebrain cellular respiration (Table 1).
Hepatotoxicities are the leading cause of drug adverse events. Therefore, it is imperative to develop adequate biomarkers that can evaluate drug candidates for their mechanisms of action and toxicity. These markers should be sensitive for detecting the earliest phases of disease. The results here show liver cellular respiration is preserved in the presence of valproate (Figure 5 and Table 1). This effect was noted despite morphologic liver abnormalities induced by the drug (Figure 6). Thus, it appears that the studied biomarkers (cellular bioenergetics, GSH and caspase activities) are not sensitive for early detection of liver injuries due to valproate.
The nucleoside reverse transcriptase inhibitor (NRTI) azidothymidine is an essential therapy for treatment of human immunodeficiency virus-1 (HIV-1). The drug is well known to induce mitochondrial damages in several organs including the heart and liver [19-20]. The results here show the drug has no significant effects on hepatocyte respiration (Table 1).
Selecting appropriate compounds for testing is a significant challenge. The candidate must be active in vitro, relatively stable for the length of experiment, has efficient cellular uptake, has rapid mode-of-action, and target organs that are amenable to testing. There and other required criteria currently limit the applications of this analytical tool. Future modifications of the procedure, however, may allow easier implementations.
In summary, the above findings justify using cellular bioenergetics to monitor the effects of drugs in targeted and off-targeted tissues. Studies are needed to assess the impact of drug dosing, exposure time, and combinations of agents on cellular respiration in various organs. Furthermore, adverse events of some drugs may require accumulative dosing or genetic susceptibility. These prospects could be tested in the future, using repetitive dosing and certain mouse strains.
Acknowledgment
The authors are grateful to Mr. Jose Kochiyil, Mr. Thachillath Pramathan, and Dr. Bayan Al-Dabbagh for performing oxygen measurements.
Author Contribution
Suleiman Al-Hammadi, Ali Alfazari and Abdul-Kader Souid designed the study, carried out the analysis, interpreted the data, and drafted the manuscript. Sami Shaban programmed the oxygen analyzer and performed data analysis. All authors have read, edited and approved the final manuscript.
Funding
This research was supported by grants from the Emirates Foundation (21M049) and from the UAE University (NRF 31M096).
References
- Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today. 2007; 12: 777-785.
- Hynes J, Marroquin LD, Ogurtsov VI, Christiansen KN, Stevens GJ, Papkovsky DB, et al. Investigation of drug-induced mitochondrial toxicity using fluorescence-based oxygen-sensitive probes. Toxicol Sci. 2006; 92: 186-200.
- Alfazari AS, Al-Dabbagh B, Almarzooqi S, Albawardi A, Souid AK. A preparation of murine liver fragments for in vitro studies: liver preparation for toxicological studies. BMC Res Notes. 2013; 6: 70.
- Alfazari AS, Al-Dabbagh B, Almarzooqi S, Albawardi A, Souid AK. Bioenergetic study of murine hepatic tissue treated in vitro with atorvastatin. BMC Pharmacol Toxicol. 2013; 14: 15.
- Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011; 18: 2995-3014.
- Luo J, Sobkiw CL, Hirshman MF, Logsdon MN, Li TQ, Goodyear LJ, et al. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006; 3: 355-366.
- Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010; 10: 267-277.
- Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010; 1: 530-543.
- Kitamura Y, Koide M, Akakabe Y, Matsuo K, Shimoda Y, Soma Y, et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J Biol Chem. 2014; 289: 2788-2800.
- McLean BA, Zhabyeyev P, Pituskin E, Paterson I, Haykowsky MJ, Oudit GY, et al. PI3K inhibitors as novel cancer therapies: implications for cardiovascular medicine. J Card Fail. 2013; 19: 268-282.
- Sellers WF. Inhaled and intravenous treatment in acute severe and life-threatening asthma. Br J Anaesth. 2013; 110: 183-190.
- Morgan DJ, Paull JD, Richmond BH, Wilson-Evered E, Ziccone SP. Pharmacokinetics of intravenous and oral salbutamol and its sulphate conjugate. Br J Clin Pharmacol. 1986; 22: 587-593.
- Treatment of preeclampsia and eclampsia [1-3,12,13 ]. 2014.
- Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013; 46: 1323-1338.
- Vitins AP, Kienhuis AS, Speksnijder EN, Roodbergen M, Luijten M, van der Ven LT, et al. Mechanisms of amiodarone and valproic acid induced liver steatosis in mouse in vivo act as a template for other hepatotoxicity models. Arch Toxicol. 2014.
- Jafarian I, Eskandari MR, Mashayekhi V, Ahadpour M, Hosseini MJ. Toxicity of valproic acid in isolated rat liver mitochondria. Toxicol Mech Methods. 2013; 23: 617-623.
- Sitarz KS, Elliott HR, Karaman BS, Relton C, Chinnery PF, Horvath R, et al. Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Mol Genet Metab. 2014; 112: 57-63.
- Visudtibhan A, Bhudhisawadi K, Vaewpanich J, Chulavatnatol S, Kaojareon S. Pharmacokinetics and clinical application of intravenous valproate in Thai epileptic children. Brain Dev. 2011; 33: 189-194.
- Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov. 2003; 2: 812-822.
- Liu Y, Shim E, Nguyen P, Gibbons AT, Mitchell JB, Poirier MC. Tempol protects cardiomyocytes from nucleoside reverse transcriptase inhibitor-induced mitochondrial toxicity. Toxicol Sci. 2014; 139: 133-141.
- Fillekes Q, Kendall L, Kitaka S, Mugyenyi P, Musoke P, Ndigendawani M, et al. Pharmacokinetics of zidovudine dosed twice daily according to World Health Organization weight bands in Ugandan HIV-infected children. Pediatr Infect Dis J. 2014; 33: 495-498.
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of Neuroscience. 1997; 17: 2921-2927.
- Alsamri MT, Al-Hammadi S, Shaban S, Pramathan T, Kochiyil J, Alshamsi A, et al. Impaired forebrain cellular bioenergetics following acute exposure to ammonia. J Clin Toxicol. 2014; 4: 189.
- Lo LW, Koch CJ, Wilson DF. Calibration of oxygen-dependent quenching of the phosphorescence of Pd-meso-tetra (4-carboxyphenyl) porphine: A phosphor with general application for measuring oxygen concentration in biological systems. Anal Biochem. 1996; 236: 153-160.
- Shaban S, Marzouqi F, Al Mansouri A, Penefsky HS, Souid AK. Oxygen measurements via phosphorescence. Comput Methods Programs Biomed. 2010; 100: 265-268.
- Tao Z, Goodisman J, Penefsky HS, Souid AK. Caspase activation by anticancer drugs: the caspase storm. Mol Pharm. 2007; 4: 583-595.
- Tao Z, Goodisman J, Souid AK. Oxygen measurement via phosphorescence: reaction of sodium dithionite with dissolved oxygen. J Phys Chem A. 2008; 112: 1511-1518.