
Review Article
Austin J Comput Biol Bioinform. 2014;1(2): 5.
Combination of Molecular Modeling and Quantum Mechanical Studies to Understand Quinolone Resistance Mechanism of Mycobacterium Tuberculosis
Joginipelli S1, Melapu VK1 and Darsey J2*
1Department of Bioinformatics, University of Arkansas at Little Rock, USA
2Department of Chemistry, University of Arkansas at Little Rock, USA
*Corresponding author: Darsey J, Department of Chemistry, University of Arkansas at Little Rock, 2801 S. University Ave, Little Rock, AR 72204, USA
Received: August 04, 2014; Accepted: September 12, 2014; Published: September 17, 2014
Abstract
The increasing emergence of multiple and extensive drug resistant tuberculosis poses a significant threat to effective control of tuberculosis (TB) globally. Resistance is a serious threat in the battle against the treatment and eradication of Tuberculosis. The continuing rise in tuberculosis incidence and the problem of drug resistance strains have prompted the research on developing new drug candidates and understanding the mechanism of drug resistance. Fluoroquinolone resistance poses a significant threat to the effective control of Multiple Drug Resistant Tuberculosis (MDR-TB) and Extensive Drug Resistant Tuberculosis (XDR-TB). Ciprofloxacin, Levoflaxacin, Gatifloxacin, and Moxifloxacin represent the important class of fluoroquinolones that inhibit mycobacterial DNA gyrase enzyme. We used combination of molecular modeling and quantum mechanical calculations to understand a compare the binding affinities and electronic structural properties of fluoroquinolones. Molecular docking studies were performed on DNA gyrase, with quinolones and the binding affinities were ranked with scoring functions. Three different docking modes were used to compare the binding affinities; moxifloxacin showed high binding affinity in all the three docking modes and showed hydrogen bonding and hydrophobic interactions with the active site of the target protein. Semi-empirical quantum mechanical Hartree-Fock formalism is used to understand the electronic structural properties of quinolones. Reactivity of the molecules was compared from electronic structural perspective; moxifloxacin showed high reactivity with more number of delocalized electrons.
Keywords: Fluroquinolone; Multiple drug resistant tuberculosis; Extensive drug resistant tuberculosis; Molecular modeling; Quantum mechanical studies; Hartree-Fock
Introduction
Tuberculosis is alarming across the globe, especially in developing countries. Drug resistance in mycobacterium tuberculosis poses a significant and serious threat to the effective treatment of Tuberculosis. Multiple Drug Resistant Tuberculosis (MDR-TB) is defined as resistance to Isoniazid (INH) and Rifampicin (RPM) drugs [1]. Second line drugs i.e. quinolones are used to treat multiple drug resistance in mycobacterium. Quinolones are considered as one of the most effective second-line drugs that are used to treat MDR-TB. Mycobacterium tuberculosis develops resistance to quinolones after prolonged exposures to the antibiotics, and emerges as extensive drug resistant strain. Extensive drug resistant strain of mycobacterium is not only resistant to INH and RPM, but also resistant to quinolones as well as one of the three injectable drugs such as Capreomycin, Kanamycin, and Amikacin [2]. Quinolones target the DNA gyrase enzyme, a type II topoisomerase present in m. tuberculosis and cause microbial death by inhibiting DNA gyrase. DNA gyrase regulates DNA supercoiling and nucleic acid tangling in mycobacterium [3]. Although there are many different mechanisms involved in development of resistance in mycobacterium, the spontaneous mutations in its gene sequence, Drug efflux systems in bacterium and its cell wall are considered as important ways. Mutations in gyrA and gyrB genes that encode DNA gyrase leads to development of quinolone resistance. DNA gyrase is a tetrameric A2B2 protein; a subunit carries the DNA breakage and reunion active site, where as the B subunit promotes ATP hydrolysis [4].
Mechanism of Action of Quinolones
Fluoroquinones represent an important class of antimicrobial which work through inhibition of DNA gyrase. Bacterial DNA gyrase (topoisomerase II) and topoisomerase IV are required for DNA synthesis [5]. Inhibition of DNA gyrase blocks relaxation of supercoiled DNA; relaxation is a requirement for transcription and replication in mycobacterium. Inhibition of topoisomerase IV is thought to interfere with separation of replicated chromosomal DNA [6,7]. In path of evolution due to long exposure to antibiotics the bacterium develops mutations in its gene sequence the can lead to resistance to quinolones.
Computational details and methodology
Molecular docking studies were performed DNA gyrase with four different quinolones, ciprofloxacin, levofloxacin, gatifloxacin, and moxifloxacin to understand the binding affinity of all the quinolones with DNA gyrase. Semi-empirical quantum mechanical calculations were performed on all the quinolones to know the electronic structural properties of all the drug molecules. The structure of DNA gyrase of Mycobacterium tuberculosis is retrieved from Protein Data Bank (PDB). The chemical structure of ciprofloxacin, levofloxacin, moxifloxacin and gatifloxacin were obtained from Pubchem database and Chemdraw ultra software is used for chemical structures. Molecular docking was performed with SYBYL x 2.0 [8], and results were analyzed with different scoring functions. Comparative molecular docking was done with different docking modes such as surflex-dock-screen, surflex-geom dock and surflex-geomx dock. Surflex-dock-screen mode is considered as fastest, and the spin density of 3 with rigid conformations is used to get the docking score. Spin density of 6 is used in surflex-geom dock and spin density of 9 is used in surflex-geomx dock. The accuracy is highest in surflex-geomx, with dynamic conformations and the results were analyzed with different scoring functions. Total score, crash score, polar score, D score, PMF score, G score, chem score, and Cscore were calculated for each docking mode. We used all different scoring functions such as force-field, empirical, and knowledge based methods. We analyzed the results with Cscore. Cscore is considered as most accurate and it incorporates multiple scoring functions. We have also compared the interaction of quinolones with DNA gyrase of staphylococcus to understand the wide range activity of the antibacterial drugs such as ciprofloxacin, levofloxacin, moxifloxacin and gatifloxacin. Comparative molecular docking was done with different docking modes such as surflex-screen-dock, surflex-geom dock, and surflex-geomx dock to understand the binding affinities of quinolones with the DNA gyrase of staphylococcus. The structure of DNA gyrase of staphylococcus was obtained from Protein Data Bank and molecular docking was performed with SYBYL x 2.0.
In addition to molecular docking studies, we performed quantum mechanical calculations on quinolones to understand the electronic structural properties. Semi-empirical Hartree-Fock formalism with 6-311G is used for these calculations to generate the Highest Occupied Molecular Orbitals (HOMO), Lowest Unoccupied Molecular Orbitals (LUMO), and contour plots. Gaussian 09 is used to calculate the total energy, dipole moment, HOMO surface and LUMO surface [9]. Gauss-view is used to view the results in different formats [10,11].
Results and Discussion
Molecular docking
Molecular docking results obtained from surflex screen dock mode, geom. mode and geomx mode shows; moxifloxacin has high binding affinity indicated with high cscore and total score and is showing hydrogen bonding with the target protein DNA gyrase. The next high binding affinity was shown by ciprofloxacin, indicating its role in fighting against the quinolone drug resistance. All the drug molecules showed hydrogen bonding and hydrophobic interactions with the target protein. The graphs were drawn for all the docking modes with cscore and total scores for all the quinolones to compare the binding affinity of the molecules with the protein target. Figure 1, 2 & 3 shows cscores and total scores of all quinolones in graphical format. The cscores and total scores obtained for ciprofloxacin, levofloxacin, moxifloxacin and gatifloxacin with surflex screen mode are shown here in the graphical format. In this docking mode moxifloxacin shows high cscore as well as high total score indicating its role in binding to DNA gyrase.
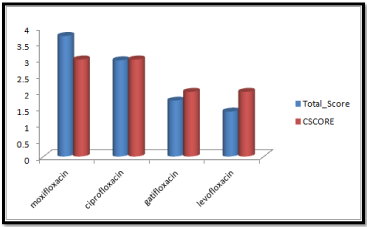
Figure 1: Cscore and total scores of quinolones with surflex screen docking mode.
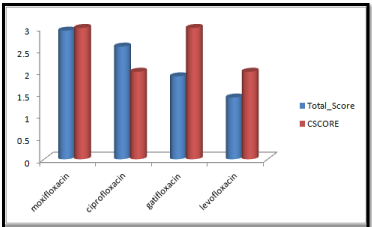
Figure 2: Cscore and total scores of quinolones with surflex geom docking mode.
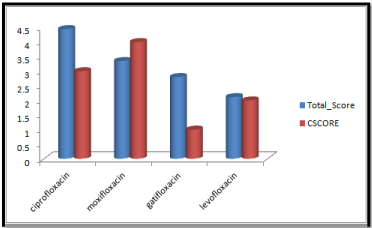
Figure 3: Cscore and total score of quinolones with surflex-geomX docking mode.
Moxifloxacin shows high score and total score in surflex geom docking mode, indicating its role in binding to DNA gyrase.
Ciprofloxacin showed high total score, and moxifloxacin showed high cscore with surflex geomx docking mode, indicating their possible role in binding to DNA gyrase.
All molecules showed hydrogen bonding with the active site. Evaluations of docking results depend mostly on interaction of molecules with the target active site of the protein. Here all the molecules showed hydrogen bonding with the receptor protein and moxifloxacin and ciprofloxacin showed more number hydrogen bonds. Figure 4 shows hydrogen bonding of ciprofloxacin, Figure 5 shows hydrogen bonding of gatifloxacin, Figure 6 shows hydrogen bonding of levofloxacin and Figure 7 shows hydrogen bonding of moxifloxacin with active site of target protein.
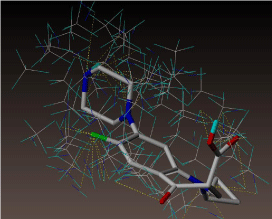
Figure 4: Ciprofloxacin showing Hydrogen bonding with active site of DNA gyrase. Here in the figure; ciprofloxacin is showed as capped stick model, active site of the DNA gyrase is shown in lines and the yellow dotted lines indicate the hydrogen bonding of the ciprofloxacin with active site of the protein.
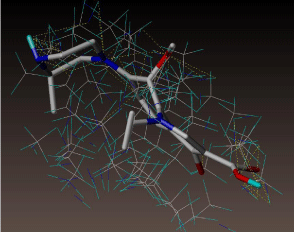
Figure 5: Gatifloxacin showing Hydrogen bonding with active site of DNA gyrase. Here in the figure; gatifloxacin is showed as capped stick model, active site of the DNA gyrase is shown in lines and the yellow dotted lines indicate the hydrogen bonding of the gatifloxacin with active site of the protein.
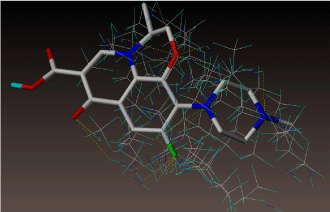
Figure 6: Levofloxacin showing Hydrogen bonding with active site of DNA gyrase. Here in the figure; levofloxacin is showed as capped stick model, active site of the DNA gyrase is shown in lines and the yellow dotted lines indicate the hydrogen bonding of the levofloxacin with active site of the protein.
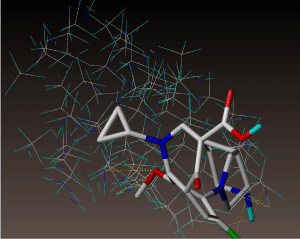
Figure 7: Moxifloxacin showing Hydrogen bonding with active site of DNA gyrase. Here in the figure; moxifloxacin is showed as capped stick model, active site of the DNA gyrase is shown in lines and the yellow dotted lines indicate the hydrogen bonding of the moxifloxacin with active site of the protein.
Docking results obtained from the interaction of quinolones with DNA gyrase of staphylococcus shows, high binding affinity of moxifloxacin when compared to other quinolones. Ciprofloxacin, levofloxacin, gatifloxacin and moxifloxacin showed hydrogen bonding with the active site of the DNA gyrase, and moxifloxacin showed high Cscore indicating its high binding affinity. Moxifloxacin showed high Cscore in all different docking modes, indicating its antibacterial activity against staphylococcus. Cscore and total scores of all quinolones in different docking modes were shown in Figure 8, 9 & 10.
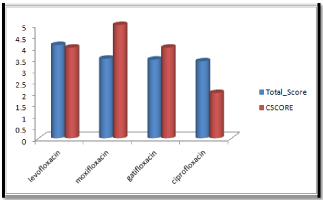
Figure 8: Cscore and total scores of quinolones with surflex screen docking mode, moxifloxacin showing high cscore with active site of DNA gyrase of staphylococcus.
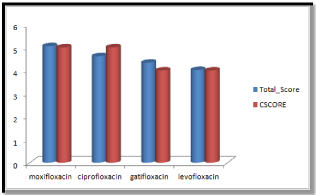
Figure 9: Cscore and total scores of quinolones with surflex geom docking mode with active site of DNA gyrase of staphylococcus.
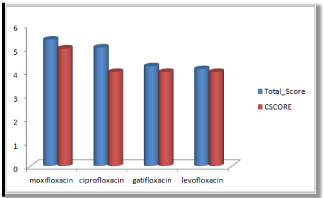
Figure 10: Cscore and total scores of quinolones with surflex geomX docking mode with active site of DNA gyrase of staphylococcus.
Quantum mechanical calculations
Theoretical calculations for conformers ciprofloxacin, levofloxacin, gatifloxacin and moxifloxacin were performed with Hartree-fock formalism and 6-311G basis set. We generated surfaces with highest occupied molecular orbitals and lowest unoccupied molecular orbitals and contours were generated to understand the reactivity of the molecule. HOMOs (Highest Occupied Molecular Orbitals) and LUMOs (Lowest Unoccupied Molecular Orbitals) determine the way the molecule interacts with other species. The frontier orbital gap helps characterize the chemical reactivity of molecule. A molecule which have more orbital gap is less polarized and less chemically reactive [12].
Figure 11 shows 3D visualization of ciprofloxacin and gatifloxacin and Figure 12 shows visualization of levofloxacin and moxifloxacin with gauss-view. According to frontier orbital theory the electro negative regions of molecule having high delocalized electron is highly reactive and participates in a reaction. HOMO and LUMO surfaces of the ciprofloxacin molecules are shown in Figure 13, the electro negative regions with highly delocalized electrons are shown in red and electro positive regions with no delocalized electrons. Ciprofloxacin shows delocalized electrons all over the surface of the molecule indicating its reactivity.
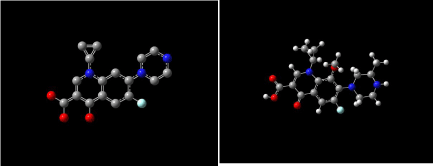
Figure 11: Ciprofloxacin and gatifloxacin visualized with gauss-view.

Figure 12: Levofloxacin and moxifloxacin with gauss-view.
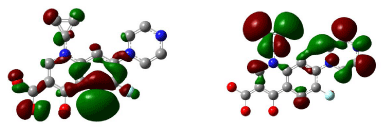
Figure 13: HOMO (Highest Occupied Molecular Orbitals) and LUMO (Lowest Unoccupied Molecular Orbitals) of ciprofloxacin.
Figure 14, 15, 16 shows HOMO, LUMO surfaces of gatifloxacin, levofloxacin and moxifloxacin respectively. From the HOMO, LUMO figures; we can say ciprofloxacin and moxifloxacin are highly reactive than gatifloxacin and levofloxacin. Ciprofloxacin and moxifloxacin have electron negative delocalized electrons all over the surface of the molecule indicating high reactivity than the other molecules.
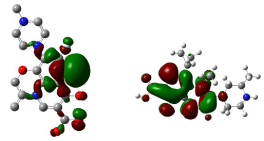
Figure 14: HOMO (Highest Occupied Molecular Orbitals) and LUMO (Lowest Unoccupied Molecular Orbitals) of gatifloxacin.
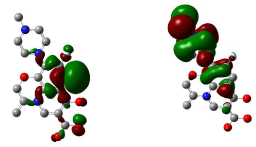
Figure 15: HOMO (Highest Occupied Molecular Orbitals) and LUMO (Lowest Unoccupied Molecular Orbitals) of Levofloxacin.
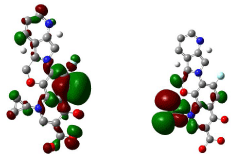
Figure 16: HOMO (Highest Occupied Molecular Orbitals) and LUMO (Lowest Unoccupied Molecular Orbitals) of moxifloxacin.
The contour plots were generated for all the molecules and compared to know the reactivity of the molecules. For a nucleophilic substitution reaction, an amount of energy equal to shielded potential energy surface is required [13-15]. The contour plots for ciprofloxacin, gatifloxacin, levofloxacin and moxifloxacin are shown in Figure 17, 18, 19 & 20. From contour plots we can say except for the oxygen molecule inside the structure of the molecules all the other oxygen atoms were not in closed contours indicating a nucleophilic reaction; all quinolones seems to be having high nucleophilic substitution reactivity.
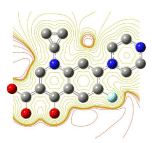
Figure 17: Contour plot of ciprofloxacin.
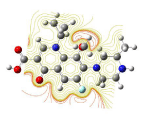
Figure 18: Contour plot of gatifloxacin.
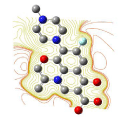
Figure 19: Contour plot of Levofloxacin.
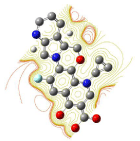
Figure 20: Contour plot of moxifloxacin.
Conclusions
This paper reports a comprehensive analysis of four different quinolones; ciprofloxacin, gatifloxacin, levofloxacin and moxifloxacin with molecule modeling and quantum mechanical studies. The analysis of molecular docking results in view of docking scores and binding affinities, moxifloxacin showed high binding affinity and hydrogen bonding, and next high binding affinity was shown by ciprofloxacin and showed more hydrogen bonds. Moxifloxacin is an 8-methoxyquinolone compound with activity against a wide range of bacteria, and can serve as a potential drug candidate for tuberculosis. The antibacterial activity was evaluated with molecular docking results obtained from both mycobacterium and staphylococcus. Quantum mechanical calculations confirmed the high reactivity of moxifloxacin with more number delocalized electron. Reactivity of molecule reflects the susceptibility of a substance towards a specific chemical reaction. This kind of quantum mechanical calculations will play a key role in designing new molecules with high reactivity and can be useful in understanding biological systems from quantum mechanical perspective. We have discussed the nucleophilic and electrophilic substitution reactions with the help of contour plots, helps in understanding the molecules in new dimensions. This work shows the need of extensive study of electronic structural properties of potential drug molecules as well as understanding the molecular mechanism of protein and ligand interactions at the active site of the target protein. This paper will encourage the need of quantum mechanical calculations on drug molecules by experimentalists who are interested in electronic as well as vibrational aspects of drug molecules.
Acknowledgement
TFigurehis project was supported by grants from the National Center for Research Resources (5P20RR016460-11) and the National Institute of General Medical Sciences (8 P20 GM103429-11) from the National Institutes of Health.
References
- Multiple Drug Resistant Tuberculosis (MDR-TB) and Extensive Drug Resistant Tuberculosis (XDR-TB), World Health Organization. 2014.
- Piton J, Petrella S, Delarue M, André-Leroux G, Jarlier V, Aubry A, et al. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS One. 2010; 5: e12245.
- Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC, et al. Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol. 2006; 8: 97-111.
- Lau RW, Ho PL, Kao RY, Yew WW, Lau TC, Cheng VC, et al. Molecular Characterization of Fluoroquinolone Resistance in Mycobacterium tuberculosis: Functional Analysis of gryA Mutation at Position 74. Antimicrobial Agents and Chemotherapy. 2011; 55: 608-614.
- Jansen G, Barbosa C, Schulenburg H. Experimental evolution as an efficient tool to dissect adaptive paths to antibiotic resistance. Drug Resist Updat. 2013; 16: 96-107.
- McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2014; 69: 292-302.
- Smith KL, Saini D, Bardarov S, Larsen M, Frothingham R, Gandhi NR, et al. Reduced virulence of an extensively drug-resistant outbreak strain of Mycobacterium tuberculosis in a murine model. PLoS One. 2014; 9: e94953.
- SYBYL-X 2.0, Tripos International. South Hanley Rd., St. Louis, Missouri, 63144, USA.1699.
- Gaussian 09, Revision D.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman J R, et al. Gaussian, Inc., Wallingford CT, 2009.
- Frisch A, Dennington II RD, Keith TD. Gauss View version 4.1 User Manual, Gaussian, Wallingford, Conn, USA. 2007.
- Jamroz MH. Vibrational Energy Distribution Analysis: VEDA 4 Program, Warsaw. 2004.
- Fleming I. Frontier Orbitals and Organic Chemical Reactions, John Wiley & Sons, New York, NY, USA. 1976.
- Bohl M, Ponsold K, Reck G. Quantitative structure-activity relationships of cardio tonic steroids using empirical molecular electrostatic potentials and semi empirical molecular orbital calculations. J Steroid Biochem. 1984; 21: 373-379.
- Lewis DF, Griffiths VS. Molecular electrostatic potential energies and methylation of DNA bases: a molecular orbital-generated quantitative structure-activity relationship. Xenobiotica. 1987; 17: 769-776.
- Kumar A, Mishra PC. "Structure-activity relationships for some anti-HIV drugs using electric field mapping." Jaournl of Molecular Structure. 1992; 277: 299-312.