
Case Report
J Dent App. 2014;1(5): 88-90.
Sturge-Weber Syndrome: A Case Report
Khandelwal S1*, Sharma S2, Patil S3 and Gupta D4
1Department of Oral Pathology & Microbiology, Dr. B R Ambedkar Institute of Dental Sciences, Patna, Bihar, India
2Practitioner, Kota, Rajasthan, India
3Department of Oral Medicine & Radiology, Jodhpur Dental College, Jodhpur, Rajasthan, India
4Department of Prosthodontics, Rajasthan Dental College, Jaipur, India
*Corresponding author: Dr. Suneet Khandelwal, 1 Ja 15, Dadabari, Kota, Rajasthan, India
Received: August 12, 2014; Accepted: September 20, 2014; Published: September 22, 2014
Abstract
Sturge-weber syndrome (SWS) is a rare, congenital, non-familial neurocutaneous disorder. A case of SWS with involvement of oral cavity is reported. The oral findings, diagnostic strategy and treatment options are discussed.
Keywords: Encephalotrigeminal angiomatosis; Oral manifestations; Sturge-Weber syndrome
Introduction
Sturge-Weber syndrome (SWS), or encephalotrigeminal angiomatosis, is a rare, congenital non-familial neurocutaneous syndrome characterized by unilateral facial cutaneous vascular malformation (nevus flammeus or port-wine stain) in association with ipsilateral leptomeningeal angiomatosis. Other symptoms associated with SWS can include eye and internal organ irregularities. Each case of SWS is unique and exhibits the characterizing findings to varying degrees [1,2].
The most apparent indication of SWS is a facial birthmark or “Port Wine Stain” (PWS) present at birth and typically involving at least one upper eyelid and the forehead. The stain, varying from light pink to deep purple, is due to an overabundance of capillaries just beneath the surface of the involved skin. Approximately 5% of patients have associated ocular involvement, mental retardation, and seizures due to the involvement of the vasculature of eye and central nervous system. A constellation of findings is called SWS [1].
In rare instances, there is an absence of a PWS. Atypical presentations such as: intracranial venous anomalies, soft tissue hypertrophy, phakomatosis pigmentovascularis, overlapped Klippel- Trenaunay syndrome (cutaneous hemangiomas, venous varicosities and soft tissue or bone hypertrophy of the affected extremities), headache and epilepsy, and an acute life-threatening event have been reported [3].
The radiographic hallmark of SWS is “tram-line” or gyriform calcifications usually involving the occipital and parietal lobes. Histologic studies have revealed that intracranial lesions of SWS display as leptomeningeal angiomatosis, and gyriform calcifications, neuronal loss, astrogliosis in underlying brain tissue.
Here, we report a case of SWS with oral changes; and discuss the clinicopathologic features, diagnostic strategy and treatment plan of the syndrome.
Case Report
A 24-year-old man presented to clinics with a history of pink to reddish discoloration of the right upper half of his face that was noticeable since birth. Discoloration is gradually progressive with swelling of upper lip of left side. Swelling on the lip varied in size time to time with no specific schedule. He also had a history of seizures that occurred once approximately 10-12 years back. He has complaints in his right eye along with slight discoloration of conjunctiva.
On general examination, the patient was in a good condition, with normal gait, built and vital signs. The physical examination revealed a port-wine nevus on the right malar region of his face involving the V1 and V2 areas of distribution of the right trigeminal nerve (Figure 1). This erythematous plaque blanches on pressure. Extra-oral examination showed swelling of right upper half of lip (Figure 2). Intra-oral examination revealed erythematous plaque involving right maxillary gingiva and opposing buccal mucosa. Massive gingival overgrowth was also seen on right maxillary half and is more prominent on buccal than palatal side. Mild deposits and stains were present all around the dentition (Figure 3). All the teeth were firm and patient had no problem in mastication. Orthopantograph findings were otherwise normal except a radiopacity in right maxillary sinus region (Figure 4). Neurologic examination was non contributory. The patient was referred for an ophthalmologic opinion. The diagnosis of glaucoma was made, and the patient was started on betaxolol (beta blocking agent) eye drops, 1 drop in each eye twice a day. There was no family history of epilepsy, mental retardation or a neurocutaneous syndrome.
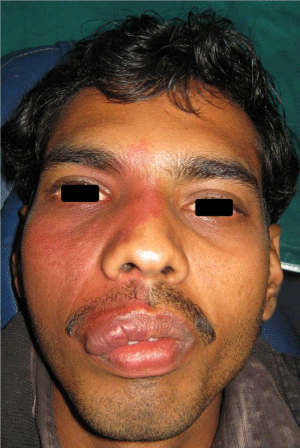
Figure 1: Port-wine stains and swelling of upper lip (involving right side of face).
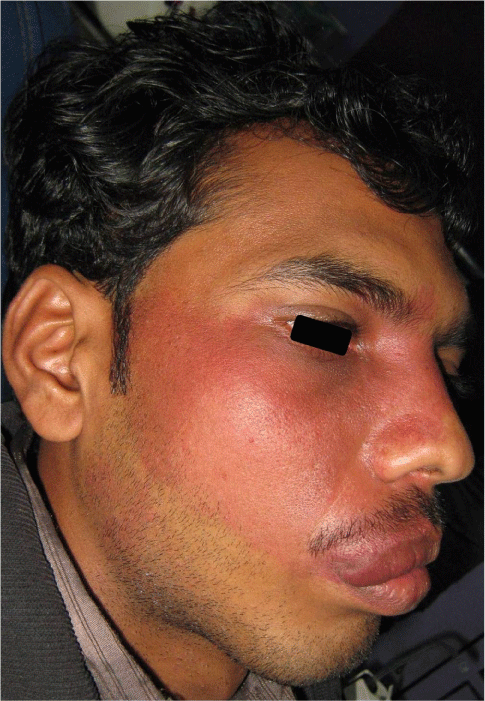
Figure 2: Extension of stains dorsally and lateral side of face.
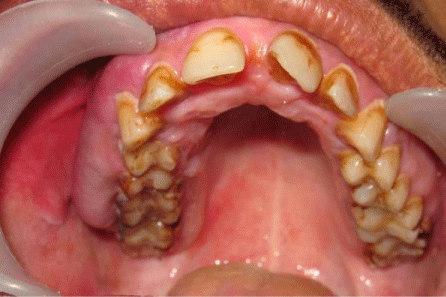
Figure 3: Intra-oral view shows gingival enlargement and erythematous buccal mucosa (right side).
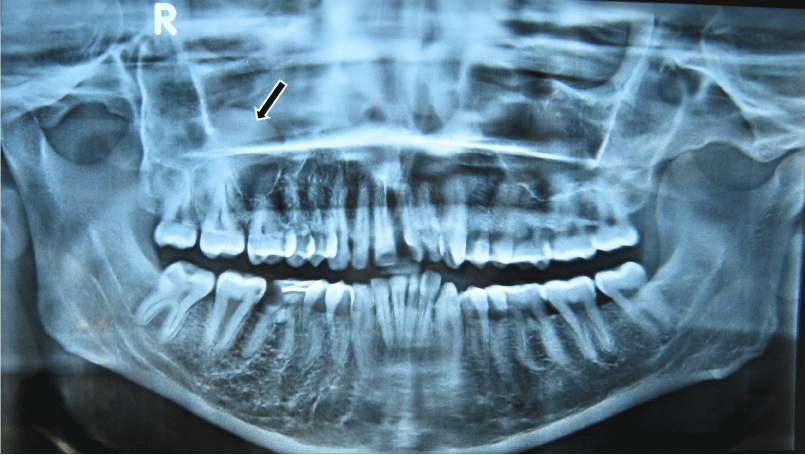
Figure 4: OPG shows mucocele of right maxillary sinus (an accidental finding).
Discussion
SWS is a congenital condition of unknown etiology which is non familial in tendency. SWS consist of a broad spectrum of pathophysiological manifestations, and all of them derive from vascular malformation affecting ipsilateral or bilateral face, leptomeninges and eyes. According to the distribution of vascular malformation, the manifestations of SWS were divided into four parts as follows. Cutaneous manifestations (unilateral facial nevus flammeus since birth, affecting the first sensory distribution of the trigeminal nerve, is the most prominent clinical finding of SWS), neurological symptoms and signs (seizure caused by hypoxia and microcirculatory is another common manifestation of SWS), ocular manifestations (glaucoma is the most common ocular complication of SWS, and presented in 30%–70% of patients) [4] and SWS may also show oral manifestations. These include unilateral vascular hyperplasia of the oral mucosa and/or gingival changes ranging from slight vascular hyperplasia to large masses, which may interfere with mouth closure.
The present case represent typical case of SWS having all four types of clinical manifestations, i.e. cutaneous (port wine stains affecting V1 and V2 areas of right trigeminal nerve), neurological (had a history of seizures in childhood), ocular (glaucoma) and oral manifestations (unilateral gingival growth and erythematous stains involving mucosa).
CT is superior to MRI in demonstrating classic gyriform calcification, which usually involves the occipital and parietal lobes underlying the leptomeningeal angiomatosis. MRI with contrast is the preferred imaging modality for the identification of structural brain abnormalities in SWS [2,4]. Thickening of the cranium on the side of the vascular malformation is also observed.
The present case underwent only for orthopantomogram which shows radiopacity in the right maxillary sinus region suggesting mucocele in the right maxillary sinus.
Clinicians have been quite familiar with SWS. However, SWS has always been missed by pathologists, because of their limited understanding for the syndrome. On gross examination, the lesion appeared grayish yellow and abnormal vessels over the surface of the lesions were found. Microscopically, leptomeninx was thickened with abundant dilated capillary vessels and a plenty of calcification was found in underlying brain tissue. One case showed proliferation of capillary vessels and thickening of arterial wall in cerebral cortex, while two cases showed hyperplasia of glial cell in white matters, and cortex atrophy and neuronal degeneration was found in one case. In initial pathological examination, two cases were diagnosed as SWS, while the others were misdiagnosed as cerebral vascular malformation [5]. This case did not undergo any histopathological examination of lesion.
The SWS is a sporadic, congenital neurocutaneous syndrome, of which the etiology remains unclear [2]. Currently, as proximity of the portions of the ectoderm destined to form the facial skin and the parieto-occipital area of the brain, most investigators believe that the combined cutaneous and cerebrovascular lesions relate to localized primary venous dysplasia between weeks 4 and 8 of pregnancy [6]. Some neurologists suggested that in SWS the vascular plexus fails to regress, as is normal during the ninth week [2]. The absence of normal leptomeningeal vessels, obstruction occurring within the vascular malformation can cause stasis, ischemia, hypoxia, and decreased neuronal metabolism, especially during seizures.
Ischemia is associated with severe physiologic changes, including hypertrophy of the choroid plexus, increased capillary permeability, hypoxia in the adjacent tissue, alterations in pH, calcium deposition, cerebral atrophy, and disruption of the blood-brain barrier [6].
SWS is referred to as complete when both CNS and facial angiomas are present and incomplete when only one area is affected without the other. The Roach Scale is used for classification, as follows: According to clinical manifestations of effected patients, SWS has been sub classified into four groups: type I, both facial and leptomeningeal angiomas, glaucoma possible (classic SWS); type II, facial angioma without evidence of intracranial disease; type III, isolated leptomeningeal angioma and possibility of glaucoma; and type IV, SWS associated with other disorders, such as tuberous sclerosis [7]. When cutaneous signs are absent, as in type III patients, the diagnosis can be made only if leptomeningeal angiomatosis is demonstrated by imaging or histological examination.
Stoke-like episodes and seizures may further exacerbate ischemic and metabolic compromise of brain tissue resulting in further injury. Therefore, prevention of seizure appears to be at the heart of the treatment for SWS. In patients whose seizures are difficult to control, neurosurgical procedures such as lobectomy or hemispherectomy appear to be the best options [8].
Laser therapy for facial cutaneous vascular malformations should begin soon after diagnosis for the best results. Without laser therapy, the lesions may thicken, darken over years and even develop nodularity, in rare instances, the cutaneous vascular malformations may lead to overgrowth of the soft tissue and bone beneath the lesion [4].
Lifelong medical treatment coupled with frequent surgeries is standard management for SWS associated glaucoma. The goal is to control intraocular pressure to prevent optic nerve damage. Medications should be administered to decrease the production of aqueous fluid or promote the outflow of aqueous fluid. Trabeculectomy and goniotomy are typical surgical options [4].
For inoperable lesions, patient is advised to sleep with the head elevated at a 45° for the rest of their lives to avoid venous distention that may occur as a result of supine positioning. Dermabrasion, tattooing, lash lamp tunable dye laser therapy can be used as other treatment modalities. For correcting lip and other oral soft tissue deformity cryosurgery may be helpful [9].
The prognosis of SWS varies widely. Of the longitudinal studies published, the presence of a seizure disorder is associated with poorer clinical outcome. Bilateranial leptomeningeal angiomatosis, which is uncommon, is associated with poorer clinical outcome [10].
Conclusions
Diagnosis of SWS with classical features is not a tough task but it is difficult to diagnose a case of atypical SWS with only intracranial or facial vascular abnormalities. In any case of suspicion imaging should be performed to screen for intracranial leptomeningeal angiomatosis. In addition, follow-up visit should be emphasized particularly in infants and children.
References
- Lin DD, Gailloud P, McCarthy EF, Comi AM. Oromaxillofacial osseous abnormality in Sturge-Weber syndrome: case report and review of the literature. Am J Neuroradiol 2006; 27: 274-277.
- Di Rocco C, Tamburrini G. Sturge-Weber syndrome. Childs Nerv Syst. 2006; 22: 909-921.
- Yallapragada AV, Cure JK, Holden KR. The Sturge Weber syndrome variant with atypical intracranial findings: case report. J Child Neurol 2006; 21: 155-157.
- Thomas-Sohl KA, Vaslow DF, Maria BL. Sturge-Weber syndrome: a review. Pediatr Neurol. 2004; 30: 303-310.
- Carrasco L, Pastor A, Farina C, Martin L, Manzarbeitia F, et al. Acral arteriovenous tumor developed within a nevus flammeus in a patient with Sturge-Weber syndrome. Am J Dermatopathol 2003; 25: 341-345.
- Comi AM. Advances in Sturge-Weber syndrome. Curr Opin Neurol 2006; 19: 124-128.
- Kremer S, Schmitt E, Klein O, Vignal JP, Moret C, et al. Leptomeningeal enhancement and enlarged choroid plexus simulating the appearance of Sturge-Weber disease in a child with tuberous sclerosis. Epilepsia 2005; 46: 595-596.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol 2003; 18: 509-516.
- Suprabha BS, Baliga M. Total oral rehabilitation in a patient with portwine stains. J Indian Soc Pedod Prev Dent 2005; 23: 99-102.
- Zaroff CM, Isaacs K. Neurocutaneous syndromes: behavioral features. Epilepsy Behav 2005; 7: 133-142.