
Research Article
J Drug Discov Develop and Deliv. 2016; 3(2): 1024.
Self Micro-Emulsifying Drug Delivery System for Lymphatic Uptake of Darunavir
Bhalekar MR*, Pokale R, Bandivadekar M, Madgulkar A and Nagore P
Department of Pharmaceutics, AISSMS College of Pharmacy, India
*Corresponding author: Bhalekar MR, Department of Pharmaceutics, AISSMS College of Pharmacy, Pune, Maharashtra, India
Received: July 18, 2016; Accepted: August 24, 2016; Published: September 09, 2016
Abstract
The aim of the present study was to target Darunavir (DRV); an antiretroviral Protease Inhibitors (PI) drug to lymphatic system using Self Micro Emulsifying Drug Delivery System (SMEDDS) to increase its solubility and bioavailability. SMEDDS system compromising of Imwitor 988 as oil phase, Tween 20 and Span 20 as binary surfactant system were optimized with respect to drug solubilization, particle size, zeta potential, dispersibility, optical clarity, cloud point, in-vitro release and thermodynamic stability. The selected formulation was subjected to ex-vivo lymphatic uptake studies using everted sac method in presence and absence of lymphatic uptake blocker Pluronic-F68. In-vivo pharmacokinetic studies were performed and lymphatic transport of DRV SMEDDS was studied. SMEDDS system containing Imwitor 988 (20%) and Surfactant mix (Smix) (80%) showed maximum drug solubility with least particle size. The ex-vivo lymphatic uptake studies of DRV loaded SMEDDS in presence of lymphatic uptake blocker showed 36.69% drug permeation which increased to 64.24% in absences of lymphatic blocker, indicating the drug transport by lymphatic path. Cmax of DRV loaded SMEDDS was higher as compared to marketed tablet. Also, Pluronic F68 treated rats show lesser plasma concentration as compared to those administered with SMEDDS. The results suggest that SMEDDS is a promising drug delivery system to improve solubility and lymphatic transport of anti-HIV drug DRV.
Keywords: Structured lipid; Imwitor 988; Ternary phase; Everted sac method; Pluronic F68; Lymphatic uptake
Abbreviations
PI: Protease Inhibitor; HIV: Human Immunodeficiency Virus; AIDS: Acquired Immune Deficiency Syndrome; Pgp: Polyglycoprotein
Introduction
SMEDDS is an isotropic, thermodynamically stable mixture of oil, surfactant/co-surfactant and drug which in contact with aqueous fluids spontaneously forms oil-in-water (o/w) microemulsion. SMEDDS have attracted growing interest as promising means for the delivery of poorly water-soluble drugs. SMEDDS have gained this popularity largely due to their excellent efficiency in improving the drug solubility, increasing bioavailability and simplicity of preparation. The nano-sized droplets have very high surface to volume ratios which are able to efficiently solubilize the drug. The drug is released in a more reproducible manner which will become less dependent on the GI physiology and the fed/fasted state of the patient [1].
DRV, a non-peptide protease inhibitor, has been shown to be extremely potent against wild type HIV and active against large panel of protease inhibitor-resistant clinical isolates. It has shown a high genetic barrier to the development of antiretroviral resistance [2]. DRV suffers from poor oral absolute bioavailability (37%), the reasons can be assumed as 1) poor aqueous solubility (0.15mg/ mL) 2) substrate to Pgp which causes efflux of the absorbed drug back into the intestinal lumen and 3) substrate to cyp3A mediated metabolism resulting into inactive metabolite [3]. Its bioavailability can be increased to 82% by combining with ritonavir, which is a potent cyp3A inhibitor. In current therapeutic treatment, the DRV/ Ritonavir combination is taken as 800mg/100mg q.d. regimen in antiretroviral- naïve patients, and as a 600mg/100mg b.i.d. or 400mg/100mg q.d. regimen in antiretroviral-experienced individuals infected with HIV [4].
Any formulation strategy which can increase solubility and inhibit the other two reasons mentioned earlier and most importantly target lymphatic system have a chance to increase the bioavailability of DRV. The lymphatic system can be a target for treatment of other diseases such as AIDS and cancer. HIV, which causes AIDS, colonizes lymphoid organs such as the spleen, thymus and lymph nodes. In the early stage of infection and throughout the latent stage, the virus replicates vigorously in lymphoid organs, meaning that lymphatic drug delivery can be advantageous in the treatment of AIDS [5]. Drug absorbed into lymphatic system empty into systemic circulation thereby surpassing cyp3A metabolism. SMEDDS have been proved to increase solubility of poor soluble drugs and its ability to target lymphatic system has been well reported in past years. Myers et al. [6] reported 3% lymphatic uptake of the absorbed dose Penclomedine when formulated as an emulsion using soybean oil. Hauss et al. [7] reported 9% increased in bioavailability of Ontazolast when administered as a 20% soybean oil-in-water emulsion when compared to suspension formulation which had bioavailability less than 1%.
Gao et al. [8] investigated the impact of different amount of oil or surfactant included in self-micro emulsifying drug delivery systems on the intestinal lymphatic transport of Sirolimus. They reported absorption from oil-free formulation was mostly via the portal blood, in contrast to the SMEDDS formulations containing =25% MCT in which the lymphatic transport of sirolimus was a major contributor to oral bioavailability.
Most studies investigating lymphatic uptake using lipid, emphasizes on use of lipids having long chain fatty acids as compared to medium or short chain fatty acids. Also triglycerides are more favored as compared to mono or di-glycerides. Kiyasu et al. [9] reported that short- and medium-chain fatty acids (with a carbon chain length shorter than 12 carbon atoms) are transported to the systemic circulation by the portal blood and are not incorporated to a great extent in chylomicrons. Long-chain fatty acids and monoglycerides are re-esterified to triglycerides within the intestinal cell, incorporated into chylomicrons and secreted from the intestinal cell by exocytosis into the lymph vessels. They demonstrated cholesterol uptake by lymphatic system when cholesterol was dissolved in triglycerides with different fatty acid chain length. They reported that uptake increased as chain length of the fatty acid increased from C2 to C18. Holm et al. [10] reported lymphatic transport of halofantrine (43.3%) when dosed in SMEDDS formulated with structured triglyceride having mixture of fatty acid chain length (C8, C18) on same glycerol backbone.
Though the above findings are impressive, there are major concerns while formulating self-emulsifying/ self-micro emulsifying/ self-nano-emulsifying drug delivery using long chain fatty acids or triglycerides with long chain fatty acids as oil phase is their less solubilization capacity for drug. Secondly, they are difficult to selfemulsify and form less stable emulsion/microemulsion/nanoemulsion as compared to oils having small or medium chain fatty acids. Thus the aim of this study was to use structured lipid (Imwitor 988, having mixture of mono and diglyceride with fatty acids having chain length C8; Caprylic acid and C10; Capric acid) as oil phase to formulating self-micro emulsifying drug delivery system and evaluate its potential for lymphatic targeting of anti-retroviral drug DRV.
Materials and Methods
Materials
DRV was received as a kind gift from Lupin Research Park, Pune, India. Imwitor 988 was supplied as a gift sample from Cremer, Germany. Tween 20 & Span 20 was purchased from Lobachemie, Mumbai, India. Neusilin US2 was gifted by Fuji Chemicals, Mumbai, India and all other chemicals were procured from the local sources.
Saturation solubility studies
Saturation solubility of DRV in various oils, surfactants, co surfactants was determined by shake flask method. In this study, DRV was added in 25mg increments to 2gm of each vehicle and the mixture was mixed using cyclo mixer. The samples were equilibrated for 24 hours. Further they were centrifuged at 2,000rpm for 20 min. to separate the supernatant. Aliquots of supernatant were taken and were suitably diluted with chloroform and drug content was quantified by measuring absorbance at 263.5nm using UV spectrophotometer (Lab India 3000).
Construction of pseudoternary phase diagram
Microemulsion area was estimated by constructing pseudoternary phase diagram for specific Surfactant mix (Smix) at various ratios to oils by water titration method. Briefly surfactants were mixed in specific ratio like 1:1, 2:1, 3:1. Each of the Smix was added to oil in following ratios by weight: 1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2 and 9:1. These mixtures were then mixed using cyclomixer for 5 min and placed at 40°C for 1 hr so that an isotropic mixture was obtained. Milli-Q Water was added to each of the mixture under continuous stirring. After each increment of water, the mixtures were observed for their appearance (turbid or clear). Turbidity of the samples would indicate formation of a coarse emulsion, whereas a clear isotropic solution would indicate the formation of a micro emulsion [11]. The phase boundary was determined by observing the changes in the sample appearance from turbid to transparent to turbid. Percentage of oil, Smix and water at which clear mixture was formed were selected and the values were used to prepare phase diagram. The data were processed by CHEMIX 3.51™ software (Arne Standnes, Norway) to construct a pseudoternary phase diagram.
Preparation of liquid DRV loaded self micro-emulsifying drug delivery system
From the solubility study data and microemulsion area depicted by construction of ternary phase diagrams, oil to surfactant ratio and their concentration ranges were selected for further formulations of DRV loaded SMEDDS (Table 1). The Surfactant system (Smix) was prepared separately by mixing the selected surfactants in their chosen ratios from the microemulsion region of the phase diagram. Oil phase containing DRV (200mg) was added into the surfactant system with continuous stirring till the homogenous mixture was formed. The resulting microemulsion preconcentrate were stored for a day and inspected for any possible physical instability characterized by phase separation and drug precipitation.
![]()
Batch
Oil – Imwitor 988 (gm)
Smix (gm)
Dispersibility
% Transmittance
Cloud point �C
Drug Solubilization (mg/gm)
Droplet size (nm)
Zeta potential (mV)
Thermodynamic stability
F1
0.5
5.5
40 seconds
Grade A
99.73
62
269.2
169.87
-34.5
Stable
F2
1.5
5.5
53 seconds
Grade B
90.61
60
247.1
128.11
-32.3
Stable
F3
2.5
5.5
1min 32sec
Grade C
38.16
49
197.3
146.35
-7.5
Phase separation
F4
0.5
6.5
40 sec
Grade A
99.43
58
264.6
122.47
-33.2
Stable
F5
1.5
6.5
47 seconds
Grade A
99.91
63
288.8
91.47
-34.9
Stable
F6
2.5
6.5
1 min 24sec
Grade C
47.93
51
214.9
111.77
-14.7
Phase separation
F7
0.5
7.5
36 sec
Grade A
99.16
67
243.9
135.97
-30.1
Stable
F8
1.5
7.5
43 sec
Grade A
98.53
57
228.4
106.42
-36.5
Stable
F9
2.5
7.5
1 min 9sec
Grade C
38.37
53
166
129.72
-11.1
Phase separation
Table 1: Composition and evaluation of different SMEDDS formulations.
Evaluation of liquid DRV loaded self micro-emulsifying drug delivery system
Droplet size & Zeta-potential measurement of DRV-SMEDDS: Each DRV SMEDDS formulation equivalent to 40mg of drug was diluted with distilled water (20mL).The droplet size and zeta potential was determined by Malvern Zetasizer ZS 90 (Malvern Instruments, Worcestershire, UK), utilizing Dynamic Light Scattering (DLS) at a scattering angle of 90°.
Dispersibility test: The efficiency of self-emulsification was assessed by using a standard USP XXII dissolution apparatus 2 for dispersibility test. One milliliter of each formulation was added in 500ml of water at 37 ± 10°C. A standard stainless steel dissolution paddle was used with rotating speed of 50rpm to provide agitation. The time required for complete emulsification was measured. The invitro performance of the formulations was visually assessed using the following grading system:
- Grade A: Rapidly forming (within 1 min) nanoemulsion, having a clear or bluish appearance.
- Grade B: Rapidly forming, slightly less clear emulsion, having a bluish white appearance.
- Grade C: Fine milky emulsion that formed within 2min.
- Grade D: Dull, grayish white emulsion having slightly oily appearance that is slow to emulsify (longer than 2 min).
- Grade E: Formulation, exhibiting either poor or minimal emulsification with large oil globules present on the surface.
Spectroscopic characterization of optical clarity: The optical clarity of aqueous dispersions of D-SMEDDS formulation was measured spectrophotometrically. Composition was prepared according to the design and diluted to 100 times with distilled water. The % transmittance of the resultant dispersion was measured at 262nm, using distilled water as a reference.
Cloud point measurement: D-SMEDDS was diluted with water in the ratio of 1:250 and the sample was placed in a water bath. Temperature was increased gradually, at 5°C intervals (or at 2°C intervals when approaching the cloud point), temperature at which cloudiness occurs was taken as cloud point.
Thermodynamic stability studies: It is the thermodynamic stability which differentiates nano or microemulsion from emulsions as former has kinetic stability as compared to emulsion [12]. The thermodynamic stability study was used to evaluate the stability of formulations. In first part of the study, heating cooling cycles are used to see the stressed effect of heating and cooling on the microemulsion stability. In this study 1gm of each D-SMEDDS formulation was added to 50ml of water to form microemulsion and six cycles were performed between 0°C and at 45°C temperature with not less than 48hrs for each temperature cycle and observed for phase separation, creaming or cracking. Formulations were also centrifuged at 3500rpm for 30min. and observed for instability.
Drug solubilization capacity of formulated DRV-SMEDDS: Excess DRV was added to each formulation weighing 2gm. Each formulation was mixed on a cyclone mixture (REMI, India) for 10min. The mixtures were equilibrated for 24 hours at room temperature further these formulations were centrifuged at 2500rpm for 20min. The supernatant was diluted with chloroform. The concentration of DRV was determined by UV spectroscopy (Lab India 3000).
In- vitro drug release studies: In vitro release study was performed using a dialysis bag diffusion technique. The dialysis bags were hydrated in distilled water overnight before the experiment. Selected D-SMEDDS containing DRV equivalent to 75mg were placed in dialysis bags. The dialysis bags were tied at both ends and were placed in the basket of USP Type I dissolution apparatus. The baskets were immersed in 900mL distilled water, maintained at 37°C. The baskets were rotated at the speed of 50rpm. Sample (5mL) was collected at predetermined time intervals of 5, 10, 15, 20, 30, 45 and 60min. and replaced with similar volume of fresh dissolution media. The amount of drug in the aliquots was analyzed by spectroscopic method [13,14].
Ex-vivo DRV permeation studies with and without lymphatic blocker: To determine the lymphatic uptake of SMEDDS, everted gut sac studies using rat intestinal segments was performed under lymphatic transport blocker condition. Permission for carrying out in vivo studies was obtained from Institutional Animal Ethics Committee (IAEC) of AISSMS College of Pharmacy and their guidelines were followed in course of the studies (research proposal number CPCSEA/ IAEC/ PT-07/ 01-2K15).
Male Wistar rat was fasted overnight for 12hours prior isolation of gut sac. The animal was anesthetised using Ketamine: xylazine combination (40mg/kg: 20mg/kg), following which the abdominal cavity was accessed by excising the abdominal skin. The entire small intestinal segment was located and removed by cutting across the upper end of the duodenum and the lower end of ileum. The mesentery was separated by manual stripping. Intestine was washed carefully with normal saline (0.9%w/v NaCl) and different segments of small intestine were identified. A length of 8–10 cm was rapidly removed and gently everted over a glass rod. The everted intestine was then slipped off the glass rod and placed in a flat dish containing Krebs-Henseleit bicarbonate buffer oxygenated with O2/CO2 (95%/5%) at 37°C. The isolated everted intestinal segment was fixed between the ends of the tubes of Ex-vivo apparatus [15]. The ends of the intestine were tied in position with a thread. After placement of intestine on the perfusion apparatus, the apparatus were dipped into the dissolution media in the dissolution pan and the dissolution study was performed. The permeation studies were performed on five formulations: (1) DRV SMEDDS, (2) DRV SMEDDS + Pluronic F-68, (3) DRV + Imwitor 988, (4) DRV + Imwitor 988 + Pluronic F-68 (5) plain drug suspension. To evaluate the lymphatic uptake of the SMEDDS by the intestinal cells, everted gut sacs were incubated with lymphatic uptake inhibitor Pluronic F68 (20μg/ml) at 37°C for 1hour prior to experiment. The dissolution medium consisted of 900mL distilled water maintained at 37±0.5°C. A fresh intestinal segment was clamped to the perfusion apparatus. The drug diffused from the dialysis bag (MWCO=1200KD) into dissolution medium (mucosal side) to the serosal side (absorption compartment). Samples were taken from both dissolution media (amount released) and from perfusion tube (amount permeated) at 5, 10, 20, 30, 40, 50 and 60min and analyzed spectrophotometrically at 263.5nm after sonication for 45 minutes.
In-vivo pharmacokinetic study: Male, Wistar rats weighing 180-250grams were used for the study. The animal group studied is tabulated in (Table 2). The animals were fasted overnight (12 hours) prior to the dosing. DRV SMEDDS and marketed DRV tablet (crushed to powder) were suspended in distilled water and fed to group 1 and group 2 animals respectively with the aid of oral feeding needle [16]. In order to study the uptake of the prepared formulation by the intestinal enterocytes, pharmacokinetic studies were designed in presence of Pluronic F-68, a well-known lymph transport inhibitor. Group 3 received Pluronic F-68 saline solution 1 hour preceding, dosing of DRV. Blood samples (2ml) were withdrawn from the retroorbital plexus at 0, 2, 4, 6 and 8 hour time interval and collected in EDTA coated tubes. The blood samples were centrifuged immediately at 15,000rpm. For 10 minutes and the plasma was separated which was stored at -20°C in screw capped polypropylene tubes till the time of analysis. Analysis of the plasma samples to quantify the drug in the plasma was done using HPLC (Agilent 1100) method.
![]()
Group (n=6)
Treatment
Group I
DRV marketed tablet (10mg/kg of body weight)
Group II
DRV SMEDDS (10mg/kg of body weight)
Group III
DRV SMEDDS (10mg/kg of body weight)
with 1mg/hr infusion of Pluronic F68
Table 2: Animal groups for in-vivo pharmacokinetic study.
Result and Discussion
Saturation solubility studies
Total dose of the drug must get solubilized in selected excipients used for the preparation of SMEDDS. This helps to optimize the final volume of the formulation and to determine the loading dose of final dosage form [17].
As shown in the (Figure 1) Imwitor 988 showed highest solubilization capacity for drug at 323.05mg/gm. Drug solubility in selected surfactant showed the following order Transcutol>Tween 20>Tween 80>PEG>Mayasol>Span 20> Span 80. Low molecular volume oils and oils with short and medium fatty acid chain length tend to have more solubilizing capacity as compared to high molecular volume oils and oils with long fatty acid chain length. Molecular volume of Imwitor 988 (Glyceryl Caprylate/Caprate) is 388 cm3 [18] with fatty acid chain length of C8 and C10 as compared to 440, 485, 910, 956, 961 cm3 for Captex 355 [19], Olive oil [20], Corn oil [21] and Safflower oil [22] respectively. HLB of the surfactant prove to have positive effect on drug solubilization. Hydrophilic surfactants with exception of Transcutol showed higher solubilization capacity for DRV, as they increase the polarity of the phase to facilitate more drug solubilization. Imwitor 988 was selected as oil phase for further formulation development study. While in selection of surfactant system combination of high and low HLB surfactant was made. SMEDDS formulation with only high HLB surfactant face the danger of getting destabilize leading precipitation of the dissolved drug once dispersed in aqueous phase.
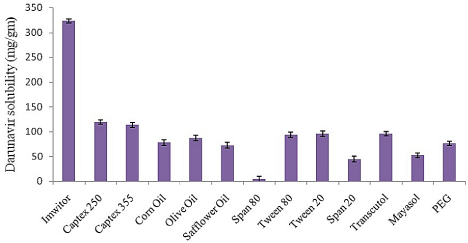
Figure 1: Solubility of DRV in various oils, surfactant and co-surfactant (n=3).
Construction of pseudoternary phase diagram
Figure 2 shows the pseudo-ternary phase diagrams with Imwitor 988 as oil phase using binary mixture of Tween’s and Span’s at different ratios as surfactants system. The colored areas indicates the clear O/W microemulsion. With Tween 80 and Span 80 as binary surfactant system maximum region of microemulsion was obtained at ratio of 1:1 (Figure 2C). Also when surfactant combination of Tween 20 and Span 20 was made maximum microemulsion area was obtained at 2:1 ratio (Figure 2E). In both the studies, microemulsion area went on increasing as the surfactant concentration increased. Above 80% surfactant concentration viscous gel phase was formed indicating the formation of bicontinoues phase.
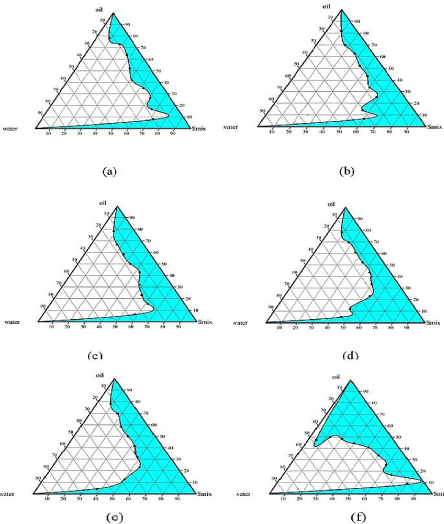
Figure 2: Pseudoternary phase diagrams indicating the microemulsion
region using Imwitor 988 as oil phase at various surfactant to co-surfactant
ratios (a) Tween 80: Span 80 (3:1) (b) Tween 80: Span 80 (2:1) (c) Tween
80: Span 80 (1:1) (d) Tween 20: Span 20 (3:1) (e) Tween 20: Span 20(2:1)
(f) Tween 20: Span 20 (1:1).
Comparing the emulsification capacity of the surfactant mixture used in the study for Imwitor 988 oil, Tween 20 and Span 20 proved to be more efficient than Tween 80 and Span 80 combination. Surfactant and co surfactant get preferentially adsorbed at the interface, reducing the interfacial energy as well as providing a mechanical barrier to coalescence. The decrease in the free energy required for the emulsion formation consequently improves the thermodynamic stability of the microemulsion formulation [23]. When combination of nonionic surfactant are used to self emulsify oil, it is observed that the chain length difference between oil and surfactant play a vital role in self-emulsification process. Less the difference in chain length of oil and surfactant more will be the spontaneous emulsification process. Tween 20 and Tween 80 have same ethoxy contained but differ only in hydrophobic chain length. Tween 20 has C12 hydrophobic chain lengths were as Tween 80 has C18, Imwitor 988 is a glyceride of medium chain fatty acid; Caprylic acid C8, Capric acid C10, our results are in line with studies reported [24].
Evaluation of D-SMEDDS
Droplet size measurement: As observed from the data of droplet size measurement, droplet size decreased as surfactant concentration increased, such results can be attributed to stabilization of the oil droplets because of the localization of the surfactant molecules at the oil-water interface which further results in interfacial disruption by enhanced water penetration into the oil droplets, mediated by the increased surfactant concentration, thus leading ultimately to the ejection of oil droplets into the aqueous phase our results are reported [25]. Further increase in surfactant concentration lead to slight increases in droplet size this might be because of formation of multi-lamellar micelles. No significant difference in the mean droplet size and polydispersity was observes when DRV SMEDDS dispersed in distilled water and simulated gastric fluid (data not shown). Lowest particle size was obtained in formulation F5 (91.5nm), F8 (106.4nm) and F6 (111.8nm). The polydispersity index was low in all batches suggesting formation of micelles with uniformity in globule size.
Zeta potential measurement: As indicated in the (Table 3) zeta potential of all the thermodynamic stable batches were more than ±30mV while the unstable batches had zeta potential of less than ±15mV. Zeta potential indicates the potential stability of the colloidal system. If the particles have low zeta potential values then there is no force to prevent the particles coming together and there is dispersion instability. A high value of zeta potential prevents particle to come close together and increases the stability of the dispersion. Particles with zeta potentials more positive than +30 mV are normally considered stable [26]. The negative charge on the droplets can be because fatty acids used in the formulation of SMEDDS. The negative charge on the surface is believed to facilitate uptake of the SMEDDS from the intestinal Payers patch, leading it to the lymphatic circulation, also it is believed to prevent entangling of the micelles in the negatively charged mucous owing to the repulsion of like charges [27,28].
![]()
Pharmacokinetic Parameters
DRV SMEDDS
Marketed
Tablet
DRV SMEDDS with lymphatic uptake blocker
Cmax
4.45 μg/ml
3.95μg/ml
3.12 μg/ml
Tmax
2 hours
2 hours
2 hours
Kele
0.099 h-1
0.053 h-1
0.108 h-1
AUCo-t
29.21 mg.hr/L
22.93 mg.hr/ L
20.95 mg.hr/ L
AUCo-8
46.71 mg.hr/ L
31.91 mg.hr/ L
37.86 mg.hr/ L
AUCt-8
17.50 mg.hr/ L
8.99 mg.hr/ L
16.90 mg.hr/ L
Half life
10.74 hours
13.08 hours
6.4 hours
Table 3: In- vivo pharmacokinetic parameters of marketed tablet, DRV SMEDDS and DRV SMEDDS with Pluronic F68 in rats.
Dispersibility test: Formulations F1, F4, F5, F7, and F8 dispersed in less than 1 minute and resulting dispersions had slight bluish appearance indicating formation of microemulsion. Formulation with Grade A and Grade B will remain as nanoemulsion when dispersed in GIT. While formulations falling in Grade C could be recommend for SEDDS formulations.
Spectroscopic characterization of optical clarity: Formulation F1, F4, F5, F7 and F8 showed % transmittance values at 99.73%, 99.43%, 99.91% 99.16%, 98.53% respectively indicating optical clarity of the dispersion system. Higher transmittances are obtained with optically clear dispersion, while cloudier dispersion will scatter more of the incident radiation, resulting in lower transmittance. Aqueous dispersions with small absorbance are optically clear and oil droplets are thought to be in a state of finer dispersion [29].
Cloud point measurement & thermodynamic stability study: The cloud point of the all batches was found to be above 37°C, suggesting formulation of a stable microemulsion of DRV at the body temperature. The formulation F1, F2, F4, F5, F7, and F8 were subjected to thermodynamic stability testing. No phase separation, creaming or cracking was observed after centrifugation in these batches.
Drug solubilization capacity of formulated DRV-SMEDDS: DRV recommended dose is 400mg/day which can be given in divided doses of 200mg twice a day thus at least 200mg of the drug has to get dissolved in one unit dose of SMEDDS formulations. All the formulation except F3 and F9 showed more than 200mg drug solubilized in 1gm of formulation.
In-vitro drug dissolution studies: The in vitro dissolution profiles of all the selected batches are shown in (Figure 3). The formulations which showed milky dispersion were not subjected to in-vitro drug dissolution study (F3, F6, and F9). SMEDDS formulations F5 and F8 showed enhanced dissolution profile as compared to other formulation. The reasons can be attribute to (a) reduced particle size which provides more surface area to release drug from solvents and thereby increases drug release rate and (b) polarity of the oil phase which itself does not diffuse through the barrier but allow drug molecules to get diffused form membrane of dialysis bag and (c) less emulsification time.
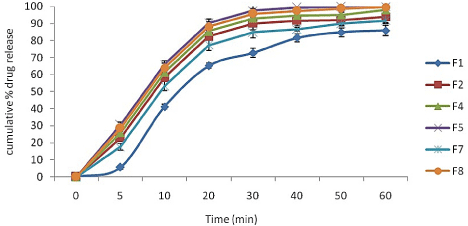
Figure 3: In-vitro release study of various formulations (n=3).
Ex-vivo DRV permeation studies with and without lymphatic blocker: The best formulation showing least globule size, highest drug loading, optical clarity and thermodynamic stability, formulation F5 was selected for further ex-vivo studies. (Figure 4) represents the % DRV permeation through everted rat intestine. The % permeation of the drug in SMEDDS formulation was 36.69% in presence of uptake blocker which increased to 64.24% in absence of lymphatic uptake blocker. Thus there was 1.74 fold increases in permeation of the drug indicating lymphatic transport of the drug. Also the drug in Imwitor 988 solution showed 1.16 fold increases in permeation in absence of lymphatic blocker indicating lymphatic transport of drug. These results can be attributed to the oil (Imwitor 988) used in the formulation of the SMEDDS, reduce droplet size, poly dispersibility, and high drug solubilization which enhance lymphatic uptake of DRV. Thus SMEDDS system formulated with glycerol of caprylic and capric acid (Imwitor 988) as oil phase has the chances of enhancing the lymphatic uptake of the drug and increase its bioavailability which was further proved by in-vivo pharmacokinetic study.
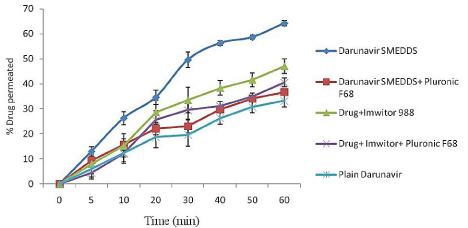
Figure 4: % Permeability of DRV through rat intestine (everted sac method)
with and without lymphatic uptake blocker (n=3).
In-vivo pharmacokinetic study: Evaluation of the in-vivo effectiveness and lymphatic transport of the SMEDDS was executed by performing in-vivo pharmacokinetic study on male Wistar rats. The plasma drug concentration versus time profile is represented in the (Figure 5). Pharmacokinetic parameters of the marketed tablet, formulated DRV SMEDDS (F5) and DRV SMEDDS with Pluronic F68 are summarized in the (Table 3). Compared to marketed tablet, a significant increase in the AUC0-t, Cmax and Tmax was observed for the DRV SMEDDS. Pluronic F68 treated rats showed lesser plasma concentration of DRV as compared to that of DRV SMEDDS group (Pluronic F68 not treated). This could be attributed to Pluronicinduced blockage of intestinal lymphatic transport. Apparently, it was concluded, loading DRV in SMEDDS formulated with Imwitor 988 oil caused the lymphatic uptake there by increasing the Cmax and the AUC and eventually increasing its relative oral bioavailability to 127%.
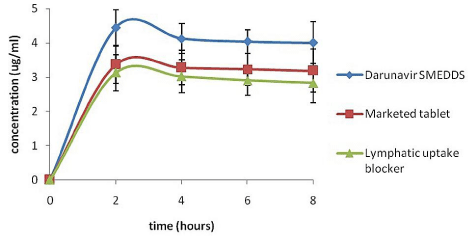
Figure 5: In vivo plasma concentrations profile of (a) Marketed tablet (b)
DRV SMEDDS (c) DRV SMEDDS with lymphatic uptake blocker (n=6, ± S.D.)
Conclusion
DRV SMEDDS system was formulated for oral administration with the aim to increase its solubility and bioavailability. Also potential of Imwitor 988 oil was accessed for lymphatic transport of drug. Selection of SMEDDS components and their concentration ranges were done on the basis of solubility study and construction of pseudo ternary phase diagram. SMEDDS prepared using Imwitor 988 as oil phase and Tween 20: Span 20 (2:1) surfactant blend had sufficient drug loading capacity, least particle size, and high optical clarity, cloud point and emulsify rapidly in aqueous media. Ex-vivo permeation and in-vivo pharmacokinetic studies in presence and absence of lymphatic uptake blocker demonstrate uptake of D-SMEDDS by lymphatic route. Thus our study highlighted that formulating DRV in SMEDDS using Imwitor 988 as oil, can be a potential strategy to improve bioavailability via solubility enhancement and lymphatic targeting.
References
- Talegaonkar S, Azeem A. Microemulsions: A novel approach to enhanced drug delivery. Recent patents on drug delivery & formulation. 2008; 2: 238-257.
- Laurenzanaa EM, Weisa CC, Bryanta CW, Newboldb R, Delclosa KB. Effect of dietary administration of genistein, nonylphenol or ethinyl estradiol on hepatic testosterone metabolism, cytochrome P-450 enzymes, and estrogen receptor alpha expression. Food and Chemical Toxicology. 2008; 40: 53–63.
- Vermeir M, Durand SL, Mannens G, Cuyckens F, Raoof A. Absorption, metabolism, and excretion of DRV, a new protease inhibitor, administered alone and with low-dose Ritonavir in healthy subjects. Drug metabolism and disposition. 2009; 37: 809-820.
- Lalanne M, Paci A, Andrieux K, et al. Synthesis and biological evaluation of two glycerolipidic prodrugs of didanosine for direct lymphatic delivery against HIV. Bioorg Med Chem Lett. 2007; 17: 2237- 2240.
- Gyseghem EV, Baert L. Co-administration of DRV and a new pharmacokinetic booster: Formulation strategies and evaluation in dogs. European Journal of Pharmaceutical Sciences. 2010; 41: 193–200.
- Myers RA, Stella VJ. Factors affecting the lymphatic transport of penclomedine (NSC-338720), a lipophilic cytotoxic drug: Comparison to DDT and hexachlorobenzene. International Journal of Pharmaceutics. 1992; 30: 51-62.
- Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AA, et al. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J Pharm Sci.1998; 87: 164-169.
- Gao L, Sun M, Zhai X, Xue K, Hu L,Yang X. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the Single-Pass Intestinal Perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. European journal of pharmaceutical sciences. 2011; 43: 132-140.
- Kiyasu JY, Bloom B, Chaikoff IL. The portal transport of absorbed fatty acids. J Biol Chem. 1952; 199: 415–419.
- Holm R, Christopher JH, Edwards GA, Mullertza A, Kristensen HG, Charman WN. Examination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in Self-Microemulsifying Drug Delivery Systems (SMEDDS) containing structured triglycerides. European Journal of Pharmaceutical Sciences. 2003; 20: 91–97.
- Deokate UA, Shinde NK, Bhingare U. Novel approaches for development and characterization of SMEDDS: Review. Int J Curr Pharm Res. 1997; 5: 5-12.
- Kallakunta, Bandari VR, Jukanti R, Veerareddy PR. Oral self emulsifying powder of Lercanidipine hydrochloride: formulation and evaluation. Powder Technol. 2005; 221: 375-382.
- Deshmukh A, Kulkarni S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev Ind Pharm. 2013; 1-11.
- Joshi M, Pathakb S, Sharma S, Patravale V. Design and in vivo pharmacodynamic evaluation of nanostructured lipid carriers for parenteral delivery of artemether Nanoject. International Journal of Pharmaceutics. 2008; 364: 119-126.
- Bhalekar MR, Kadam NM, Patil NH, Gawale NS, Madgulkar AR. Novel ion exchange resin-based combination drug delivery system for treatment of gastro esophageal reflux diseases. Brazillian Journal of Pharmaceutical Sciences. 2010; 46: 334-342.
- Inugala S, Eedara BB, Sunkavalli S, Dhurke R, Kandadi P. Solid self –nano emulsifying drug delivery syetem of DRV for improved dissolution and oral bioavailability: European Journal of Pharmaceutical Sciences. 2015; 74: 1-10.
- Puttachari S, Kalyane NV, Duttagupta S. Design and evaluation of Self-Micro Emulsifying Drug Delivery Systems (SMEDDS) of Cefuroxime Axetil. Int J Pharm Sci Rev Res. 2013; 22: 70-74.
- Chem Spider Search and share chemistry. 2016.
- Captex 355 Glyceryl Tricaprylate/ Caprate Caprylic/ Capric Triglyceride. 2006.
- Paranjpe GR, Deshpande PY. Di electric properties of some vegetable oils. Proceedings of the Indian Academy of Sciences - Section A. 1935; 1: 880-886.
- Digital Vetenskapliga Arkivet. 2016.
- Glyceryl Tricaprylate/ Caprate Caprylic/ Capric Triglyceride. 2016.
- Shukla JB, Patel SJ. Formulation and evaluation of Self Micro Emulsifying System of Candesartan Cilexetil. Int J Pharm Pharm Sci. 2010; 2: 143-146.
- Bandivadekar M, Pancholi S, Ghanekar RK, Choudhari A, Koppikar S. Self-microemulsifying smaller molecular volume oil (Capmul MCM) using non-ionic surfactants: a delivery system for poorly water-soluble drug. Drug Development and Industrial Pharmacy. 2012; 37: 883-892.
- Singh B, Bandopadhyay S, Kapil S, Singh R, Katare OP. Self-Emulsifying Drug Delivery Systems (SEDDS): Formulation development, characterization, and applications. Critical reviews in therapeutic drug carrier systems. 2009; 26: 427–521.
- Raval C, Joshi N, Patel J, Upadhyay UM. Enhanced oral bioavailability of olmesartan by using novel solid self emulsifying drug delivery system. International Journal of Advanced Pharmaceutics. 2012; 2: 82-92.
- Hussain N, Jaitley V, Florence A. Recent advances in the understanding of uptake of microparticulate across the gastrointestinal lymphatics. Advanced Drug Delivery Reviews. 2001; 50: 107-142.
- Muller H, Kovacevic AB. Solid Lipid Nanoparticles (SLN) stabilized with polyhydroxy surfactants: Preparation, characterization and physical stability investigation. Colloids and Surfaces A: Physicochem Eng Aspects. 2014; 444: 15-25.
- Chudasama A, Patel V, Nivsarkar M, Vasu K, Shishoo C. A Novel Lipid-based Oral Drug Delivery System of Nevirapine. International Journal of Pharm Tech Research. 2001; 3: 1159-1168.