
Research Article
Austin J Emergency & Crit Care Med. 2015;2(2): 1015.
Acute Lung Injury Caused by High Tidal Volume in a Rat Pneumonia Model
Julie Chi Chow1#, Wei-Lun Liu2#, Chih-Cheng Lai2, Khee-Siang Chan3, Chin-Ming Chen3,4,5*, Kuo-Chen Cheng6,7,8 and Willy Chou4,5
1Department of Pediatric, Chi Mei Medical Center, Taiwan
2Department of Intensive Care Medicine, Chi Mei Medical Center, Liouying, Taiwan
3Department of Intensive Care Medicine, Chi Mei Medical Center, Taiwan
4Chang Jung Christian University, Taiwan
5Department of Recreation and Health-Care Management, Chia Nan University of Pharmacy & Science, Taiwan
6Section of Respiratory Care, Chi Mei Medical Center, Taiwan
7Department of Internal Medicine, Chi Mei Medical Center, Taiwan
8Department of Safety Health and Environment, Chung Hwa University of Medical Technology, Taiwan #Both had an equal contribution on this paper
*Corresponding author: Chin-Ming Chen, Department of Intensive Care Medicine, Chi-Mei Medical Center, 901 Chung Hwa Road, Yang Kang City, Tainan, 71044, Taiwan
Received: January 11, 2015; Accepted: February 20, 2015 Published: February 24, 2015
Abstract
Background: To establish animal model of two-hit model for ventilator induced lung injury after pneumonia.
Methods: Male Sprague-Dawley rats (300 – 400g) were intratracheally challenged with lipopolysaccharide (LPS) as a first hit to induce lung inflammation. Rats were then randomized 24 hours later to receive mechanical ventilation as a second hit, with either an injurious strategy of high tidal volume (TV) of 22 mL/kg and zero positive end-expiratory pressure (PEEP) (high volume group, HV) or a protective strategy of low TV of 6 mL/kg with PEEP of 5 cm H2O (low volume group, LV), along with a fraction of inspired oxygen of 40 % during the experimental period. There were 4 groups (n = 8-10 rats/group): (1) HV + placebo; (2) HV + LPS; (3) LV + placebo; and (4) LV + LPS.
Results: After 4-hours of ventilator use, each group had a similar hemodynamic status (mean arterial pressures and heart rates) and arterial pH, PaCO2, and HCO3 values. However, as compared with the other groups, group 2 (HV + LPS) had lower arterial O2 and lung compliance, worse lung edema, higher total and neutrophil cell counts in lung lavage fluid, and increased lung elastance and some lung cytokines.
Conclusion: Inadequate ventilator settings may cause severe lung injury that is a complication after LPS induced pneumonia, as evidenced in our animal model by worse lung compliance, elastance, oxygenation, inflammatory cells, cytokines, and lung edema, which comply with evidence in the literature. Clinicians should be cautious regarding possible lung injury by inappropriate ventilator settings.
Keywords: Acute lung injury; Lipopolysaccharide; Mechanical ventilation; Pneumonia; Rat model
Abbreviations
ALI: Acute Lung Injury; ARDS: Acute Respiratory Distress Syndrome; BAL: Bronchoalveolar Lavage; CXCL1: chemokine ligand 1; Fio2: Fraction of Inspired Oxygen; HV: High Volume; IL: Interleukin; LPS: Lipopolysaccharide; LV: Low Volume; MAP: Mean Arterial Pressure; MV: Mechanical Ventilation; PEEP: Positive Endexpiratory Pressure; TNF: Tumor Necrosis Factor; TV: Tidal Volume; VILI: Ventilator-Induced Lung Injury; W/D: Wet To Dry
Introduction
Pneumonia that is complicated by acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS), is the leading cause of death in critically ill patients, and often requires mechanical ventilation (MV) [1]. Although MV provides essential life support, and the weaning rate may be as high as 90% in selected patients with planned extubation [2], mechanical stresses produced by MV can lead to over-distension of lung units or shear forces that are generated during repetitive opening and closing of atelectatic lung units or biotrauma. This can induce or enhance an inflammatory response that can also worsen lung injury as a ventilator-induced lung injury (VILI), with characteristics similar to those caused by ARDS [3,4]. One large multicenter trial demonstrated the importance of VILI by using ventilation with a lower tidal volume (TV; 6 mL/ kg) versus traditional TV (12 mL/kg) in which the lower TV improved survival [5]. The cytokines response can also be reversed by reinstitution of lung protective mechanical ventilation after injurious ventilator strategy [6].
Inappropriate ventilator strategies preceded by hemorrhagic shock and followed by reperfusion or intratracheal lipopolysaccharide (LPS) administration can cause a so-called “two-hit injury”; this makes the lung more susceptible to mechanical ventilation injury [7,8]. The spectrum of injuries includes disrupting endothelial and epithelial cells, increasing endothelial and epithelial permeability, neutrophil infiltration, enhanced production of inflammatory cytokines, including interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-a, and subsequent permeability pulmonary edema, hyaline membranes, and decreased lung compliance [7-9]. Clinically IL-8 levels, the equivalent of chemokine ligand 1 (CXCL1) in rats, are elevated in patients with ARDS and mechanical stress [10,11].
Thus, the aim of this study was to determine whether a large TV could augment the inflammatory response in rat lungs after they received an intra-tracheal instillation of LPS. LPS instillation to induce lung injury mimics the clinical syndromes of pneumonia and ARDS and can be used to explore any injury augmentation from large TV during MV in lungs with underlying injuries. We also investigated rats’ lung mechanics, blood gases, augmented inflammatory responses, and lung injuries.
Materials and Methods
Animal preparation
All experiments complied with protocols approved by the Institutional Animal Care and Use Committee of Chi-Mei Hospital. Briefly, LPS (Escherichia coli O55:B5, Sigma, St Louis, MO, USA) was administered intratracheally at 0.1 mg/kg into male Sprague- Dawley rats (300 – 400 g) under anesthesia (sodium pentobarbital, intra-peritoneal injection) as a first hit to induce lung inflammation. Animals were allowed to recover from anesthesia and returned to their cages with food and water provided ad libitum.
Experimental protocol
Rats were block randomized 24 hours after the intratracheal challenge with LPS and were anesthetized with sodium pentobarbital. For each rat, the right carotid artery was cannulated (Angiocath IV Catheter; 24-gauge) to monitor mean arterial pressure (MAP) and to collect samples and for blood gas measurements. A tracheostomy was performed, and a 14-gauge cannula (Angiocath IV Catheter; 2.1 x 48 mm) was inserted into the trachea. The rats were ventilated with a Servo 300 ventilator (Siemens, Sweden) using a TV of 6 ml/ kg, positive end-expiratory pressure (PEEP) of 5 cm H2O, and a respiratory rate of 50 breaths/min with a fraction of inspired oxygen (FiO2) of 40 %. A 24 gauge intravenous cannula was inserted and kept in the tail vein to infuse anesthesia or lactated Ringer’s solution to maintain adequate ventilator synchrony or blood pressure.
Thirty minutes later, rats were randomly assigned to receive mechanical ventilation at two different ventilator settings over a 4-h period. For a high volume (HV) group (injurious strategy), rats were ventilated with a TV of 22 ml/kg and zero PEEP. For a low volume (LV) group (protective strategy), rats were ventilated with a TV of 6 ml/kg with PEEP of 5cm H2O. The minute volume was maintained as constant during the entire study period by adjusting the respiratory rate to 16-18 breaths/min in the HV group. The fraction of inspired oxygen was maintained as noted above. MAP was maintained above 70 mmHg after ventilation randomization.
Rats were randomly assigned to 4 groups (10 rats/group) in a blinded manner: Group 1: HV + placebo; Group 2: HV + LPS; Group 3: LV + placebo; Group 4: LV + LPS.
Measurements of lung mechanics and blood gases and tissue sampling
Airway pressures (peak and plateau pressures) were recorded using a data acquisition system (National Instrument DAQ Card 700, Austin, TX) at a sampling rate of 200 Hz (ICU Lab, Kleis TEK Engineering, Bari, Italy). Lung elastance was calculated using the formula: (Plateau Pressure – PEEP)/TV. Blood gases were checked at the beginning of the study period and hourly until the end of the study period. Rats were humanely sacrificed at the conclusion of the experiment by sodium pentobarbital overdose. The lungs were excised via a midline sternotomy and inflated twice to an airway pressure of 30cm H2O to generate static pressure-volume curves by manually injecting 0.5 to 1cm3 aliquots of air in a stepwise manner starting at atmospheric pressure and continuing until achieving an airway pressure of 30cm H2O. This was followed by generating a deflation curve by withdrawing air using a similar step-wise approach. Volumes were maintained at each step for 6 seconds before the next air injection.
The left lungs were lavaged (bronchoalveolar lavage, BAL) for cell differentiation assays. The right upper lungs were used to measure wet to dry (W/D) lung weight ratios. The remaining part of the right lung was removed, homogenized in Triton X solution, and then centrifuged (5804R; Eppendorf, Brinkmann Instruments, Westbury, NY) at 1,000 x g at 4°C for 10 min. After centrifugation, the supernatant was collected to determine cytokine levels (IL-1, CXCL1, and TNF-a). Analysis of cytokines (IL-1, CXCL1, and TNF-a) in the plasma (n=5 for each group) and BAL fluid (n=8-10 for each group) was done in a blinded fashion by technicians using DuoSet ELISA Development kits (R & D Systems, Minneapolis, MN, USA).
Histology
Lung injury scores included alveolar collapse, alveolar hemorrhage, perivascular edema, alveolar polymorphonuclear leukocytes, membranes, and alveolar edema [7,12]. Scoring was done by a pathologist who was blinded to the experimental groups. Five regions for each specimen were examined. An injury score of 0–3 (0 = normal; 1 = mild; 2 = moderate; 3 = severe) was assigned to each region and used to calculate a total score for lung injury (n=5 for each group), which was evidenced by previous literatures [7,12].
Statistical analysis
Results are given as means ± SEM’s. Group comparisons used two-way analysis of variance (ANOVA) for repeated measures. One-way analysis ANOVA followed by a t-test was used, when appropriate. Post-hoc analysis used a Bonferroni test. A p-value of <0.05 was considered significant.
Results
After 4-hours of ventilator use, the volume of fluid that had been infused was identical (≈ 2.5 ml lactated Ringer’s solution) in all rats during the mechanical ventilation period. Each group of rats had a similar hemodynamic status (mean arterial pressures and heart rates) at baseline and during mechanical ventilation (Figure 1).
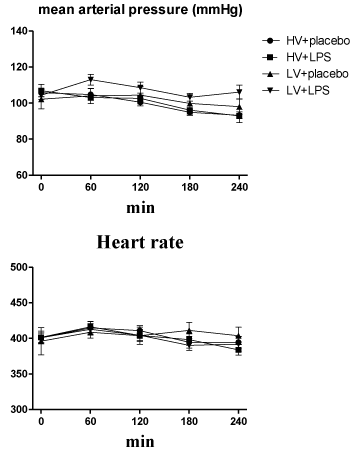
Figure 1: Time course for mean arterial pressure and heart rate during 4-hour
ventilator use after randomization. HV: High Volume; LV: Low Volume; LPS:
Lipopolysaccharide. N = 8-10 rats for each group.
Blood gas analysis and respiratory mechanics
Each group of rats also had similar arterial pH, PaCO2, and HCO3values at baseline and during randomization into the two mechanical ventilatory strategies (Figure 2). The mean PaO2 and SaO2 values at baseline were similar in all rats. However, these values were significantly lower in the HV+LPS group at 120 minutes after randomization, and the HV groups also had lower levels at the end of the study as compared with the LV groups (Figure 2). Lung elastance values at baseline were similar in all groups, but increased significantly in the HV groups compared to the LV groups at 60 minutes after randomization (Figure 3A). Similarly, the static pressure–volume curves showed that the HV groups had lower compliance, and the HV+LPS group had the worst compliance among all groups at the end of lung expansion with 30cm H2O.
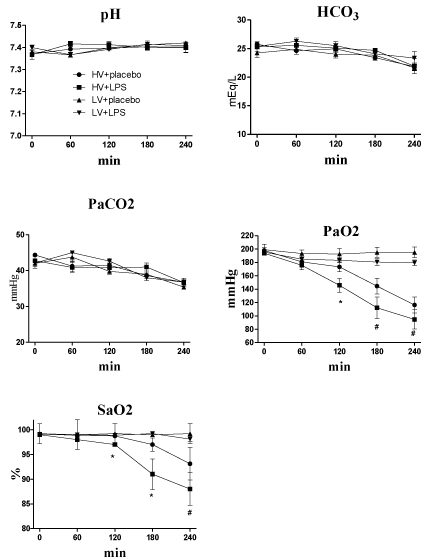
Figure 1: Blood gas results (pH, HCO3, PaCO2, PaO2 and SaO2) during
4-hour ventilator use after randomization. HV: High Volume; LV: Low Volume;
LPS: Lipopolysaccharide. *p<0.05 LPS + HV vs. other groups; # p<0.05 HV
groups vs. LV groups. N = 8-10 rats for each group.
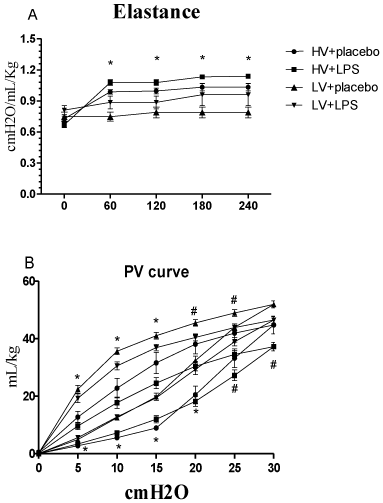
Figure 3A: (A) Elastance changes during mechanical ventilation after
randomization. *p < 0.05 HV groups vs. LV groups. (B) Static compliance
curves for the entire respiratory system at the end of the experimental
protocol. *p<0.05 HV groups vs. LV groups; # p<0.05 LPS + HV vs. other
groups. N = 8-10 rats for each group. HV: High Volume; LV: Low Volume;
LPS: Lipopolysaccharide.
PMN lung infiltration and lung edema
The total cell counts in BAL fluid were highest in the HV + LPS group, and they were significantly lower in the LV+ placebo group (0.54 ± 0.12 x 106/mL) than in the other groups (1.96 ± 0.35 x 106/ mL in HV + placebo; 2.83 ± 0.61 x 106/mL in HV + LPS; and 2.05 ± 0.24 x 106/mL in LV + LPS; all p < 0.05; Figure 4A). Similarly, the HV + LPS group had the highest percentage of neutrophils, and the LV+ placebo group had a lower level than the other groups (0% vs. 3% in HV + placebo, 81% in HV + LPS, and 74% in LV + LPS; all p < 0.05; Figure 4B).
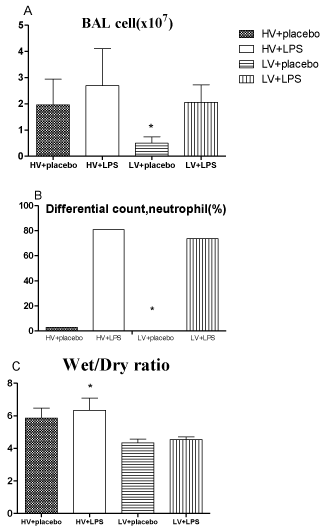
Figure 4: A. Total cell counts in lung-lavage fluids. B. Percentages of
neutrophils in lung lavage fluids. C. Lung wet-to-dry ratios. * p<0.05 vs. other
groups. N = 8-10 rats for each group. HV: High Volume; LV: Low Volume;
LPS: Lipopolysaccharide.
The lung wet-to-dry ratio was 6.30 ± 0.24 in the HV + LPS group, which was significantly higher than the other groups (5.86 ± 0.22 in HV + placebo, 4.35 ± 0.14 in LV + placebo, and 4.55 ± 0.05 in LV + LPS; all p < 0.05; Figure 4C).
Cytokine profiles
Among the ten cytokines assayed, the plasma levels of CXCL1, IL-6, and TNF-a tended to be higher in the HV + placebo group than in the other groups, although these differences were not significant (p > 0.05; Figure 5). The BAL fluid levels of CXCL1 and IL-6 were higher in the HV groups than in the LV groups, and TNF-a was higher in the HV + placebo group than the other groups (p < 0.05; Figure 5).
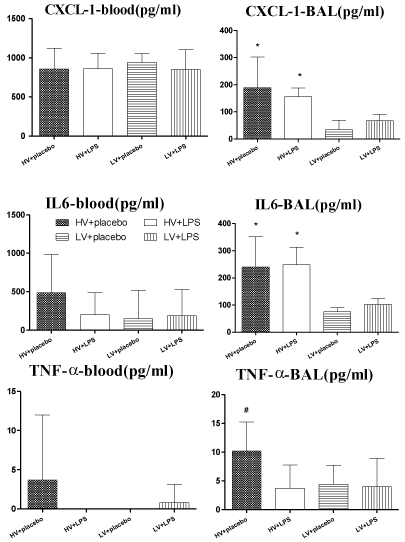
Figure 5: CXCL1, IL-6, and TNF-a levels in plasma (n=5) and lung lavage
fluid (n = 8-10) at the end of experiments. *p<0.05 HV groups vs. LV groups;
# p<0.05 HV + placebo vs. other groups. HV: High Volume; LV: Low Volume;
LPS: Lipopolysaccharide.
AbstLung injury scores and lung histologyract
The lung injury scores were higher in the HV + LPS group (9.9 ± 1.2) than in the other groups (5.3 ± 1.1 in HV + placebo, 5.8 ± 1.0 in LV + LPS, and 4.8 ± 1.5 in LV + placebo; all p < 0.05). Lung histology results showed that the HV + LPS group had aggravated alveolar collapse, perivascular and peribronchial edema, and polymorphonuclear neutrophil infiltration as compared to the placebo group (Figure 6).
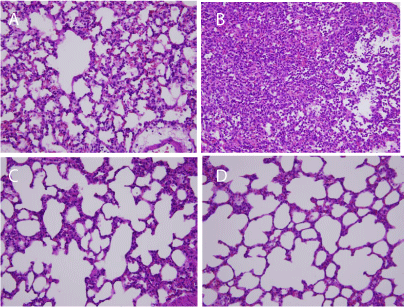
Figure 6: Representative lung histology slides (original magnification X 400).
(A) High volume + placebo, (B) high volume + LPS, (C) low volume + placebo,
and (D) low volume + LPS (n=5 in each group) . HV: High Volume; LV: Low
Volume; LPS: Lipopolysaccharide.
Discussion
This study was designed to present the physiologic and biologic profiles of experimental model used in VILI research. The main finding was that VILI in the two-hit model of LPS-injured lungs (first hit) followed by injurious mechanical ventilation (second hit) in rats caused marked degrees of lung injury (decreased PaO2 and static compliance, increased elastance and lung edema and extended lung destruction), neutrophil recruitment, and cytokine production in lung tissues. In critically ill patients with acute respiratory failure, mechanical ventilation is a lifesaving therapy; however, excessive alveolar distention and cyclic stretch associated with mechanical ventilation may by itself promote VILI. Previous studies have shown that VILI alone may not cause extensive lung injury in the normal lung, but that VILI enhances lung inflammation in pre-injured lungs [7-9,13]. Our results also showed that HV caused moderate degrees of lung injury, impaired oxygenation, worsened respiratory mechanics, enhanced inflammatory cells recruitment into the alveolar space, and increased pro-inflammatory cytokine and chemokine production and pulmonary edema. In addition, LPS-injured lungs potentiated by HV caused marked degrees of lung injury. Importantly, controls as LV groups were stable over a 4-hour period of mechanical ventilation with no obvious signs of mechanical dysfunction, inflammatory reaction, or damage to the lungs, even in the previous LPS challenge.
In experimental studies, LPS, gram-negative bacteria wall components, are often used as a surrogate for gram-negative bacteria. Previous studies reported that LPS administration into the lung could initiate monocyte/macrophage activation, neutrophil recruitment, epithelial shedding, endothelial dysfunction, and the rapid release of cytokines, chemokines, bioactive amines, and reactive oxygen species [14,15]. Marked leukocyte infiltration, alveolar edema, increased vascular permeability with enhanced plasma exudation in the lung, and severe disruption of the ultrastructure, which results in ARDSlike damage, can be caused by higher doses of LPS [16,17]. A large TV applied to LPS-instilled lungs or hemorrhagic shock followed by reperfusion, a so-called “two-hit injury,” could increase pulmonary permeability, edema, neutrophil recruitment, and cytokine release into the plasma, as demonstrated previously [7,8], and was evident in our study.
CXCL1 is a chemokine that is involved with the recruitment and activation of all leukocytes and stimulates the release of IL-1, IL-6, and TNF-a from fibroblasts and macrophages. In rats, CXCL1 is the equivalent of human IL-8. Clinically, IL-8 levels are elevated in patients with ARDS [10]. Several investigators have shown that IL-8 is released by lung cells during mechanical stretch [11]. Our observations were in agreement with these findings that CXCL1 in lung was potentiated with injurious ventilator strategy in HV group.
Tremblay et al. found large concentrations of pro-inflammatory cytokines (TNF-a, IL-6) in the BAL fluids from lungs that had been ventilated with a higher TV [18]. It was also evidenced in our study. IL-6 is a pleiotropic cytokine with a wide variety of biologic actions in acute and chronic inflammation, vascular disease, and cancer, and serum and BAL levels of IL-6 are correlated with ARDS mortality [10,19]. High tidal volumes were associated with significant increases in alveolar concentrations of IL-6, a cytokine that has been associated with inflammatory tissue injury and VILI in patients and animal models, with or without endotoxin pre-treatment [20,13].
In contrast, a previous report found that TNF-a levels were the same in the BAL fluid of LPS-administered rats with or without neutropenia [21]. This suggests that macrophages are the major source of TNF-a and that neutrophil derived TNF-a constitutes a minor portion of the total BAL fluid TNF-a. In line with the above evidence, our study presented that lung lavage cytokine levels (CXCL1, IL-6) increased with increasing TV with or without endotoxin pre-treatment, but the administration of LPS and HV to rats failed to further increase TNF-a production as compared to rats that received HV alone.
The results of our study indicate that at the onset of randomization to HV or LV, both groups had similar, relatively normal PaO2/FiO2 ratios (around 500 mmHg) and elastance, suggesting that there was little acute lung injury at that time. Although at randomization we did not perform a histological analysis, it is likely that some degree of subclinical lung injury may have been present that was not reflected by oxygenation or elastance criteria. As we hypothesized, the effects of the same underlying pulmonary injury differed depending on the type of ventilatory strategy. This was exemplified by a decreased PaO2 and compliance and increased histological lung injury score and wet-to-dry weight ratio. In general, our animal model exhibited the “two-hit injury” theory for the pathogenesis of organ injury. The first intervention (hit) primed the rat for an exaggerated response to a second intervention (hit). In these models, endotoxin is frequently used as a first intervention. In our study, endotoxin and ventilation were the two interventions that were applied in a clinically relevant and time-related manner.
The drawback of our study was the results of CXCL1, IL-6, TNF-a were not full concordance with the impairment in oxygenation and respiratory mechanics in HV + LPS group. This might be due to the limited numbers of blood cytokines (n=5) and the possibility of potential sampling mistakes on lung lavage. Further survey with more cytokines evidences (as adding IL-1 and IL-10) and more numbers of recruitment animals may improve the limitation.
Conclusion
In conclusion, our study provides evidence that rats subjected to ventilation with a large TV are more susceptible to VILI after pulmonary priming with pre-existing lung injury by intratracheal LPS administration, as we observed impaired oxygenation, worsened respiratory mechanics, enhanced inflammatory cells recruitment into the alveolar space, increased pro-inflammatory cytokine and chemokine production, and pulmonary edema. Mechanical ventilation should be carefully adjusted for patients with severe pneumonia or previous lung injury.
Funding/Support Statement
This study is supported by a Grant CMFHR9751and CMFHR10314 from the Chi-Mei Medical Center and NSC 100-2314-B-384-004 from the National Science Council in Taiwan.
References
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff Mm et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005; 353: 1685-1693.
- Cheng AC CK, Chen CM, Hsing SC, Sung MY. The outcome and predictors of failed extubation in intensive care patients-the elderly is an important predictor. International Journal of Gerontology. 2011; 5: 206-211.
- Dos Santos CC, Slutsky AS. Invited review: mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol (1985). 2000; 89: 1645-1655.
- Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. 2003; 289: 2104-2112.
- [No authors listed]. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000; 342: 1301-1308.
- Stuber F, Wrigge H, Schroeder S, Wetegrove S, Zinserling J, Hoeft A, et al. Kinetic and reversibility of mechanical ventilation-associated pulmonary and systemic inflammatory response in patients with acute lung injury. Intensive Care Med. 2002; 28: 834-841.
- Chen CM, Penuelas O, Quinn K, Cheng KC, Li CF, Zhang H, et al. Protective effects of adenosine A2A receptor agonist in ventilator-induced lung injury in rats. Crit Care Med. 2009; 37: 2235-2241.
- Lin SM, Lin HC, Lee KY, Huang CD, Liu CY, Wang CH, et al. Ventilator-induced injury augments interleukin-1beta production and neutrophil sequestration in lipopolysaccharide-treated lungs. Shock. 2007; 28: 453-460.
- Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med. 2001; 163: 1176-1180.
- Levitt JE, Gould MK, Ware LB, Matthay MA. The pathogenetic and prognostic value of biologic markers in acute lung injury. J Intensive Care Med. 2009; 24: 151-167.
- Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999; 277: L167-173.
- Ko SC, Zhang H, Haitsma JJ, Cheng KC, Li CF, Slutsky AS. Effects of PEEP levels following repeated recruitment maneuvers on ventilator-induced lung injury. Acta Anaesthesiol Scand. 2008; 52: 514-521.
- Schreiber T, Hueter L, Schwarzkopf K, Hohlstein S, Schmidt B, Karzai W, et al. Increased susceptibility to ventilator-associated lung injury persists after clinical recovery from experimental endotoxemia. Anesthesiology. 2006; 104: 133-141.
- Hudson AR, Kilburn KH, Halprin GM, McKenzie WN. Granulocyte recruitment to airways exposed to endotoxin aerosols. Am Rev Respir Dis. 1977; 115: 89-95.
- Lin SM, Frevert CW, Kajikawa O, Wurfel MM, Ballman K, Mongovin S, et al. Differential regulation of membrane CD14 expression and endotoxin-tolerance in alveolar macrophages. Am J Respir Cell Mol Biol. 2004; 31: 162-170.
- Esbenshade AM, Newman JH, Lams PM, Jolles H, Brigham KL. Respiratory failure after endotoxin infusion in sheep: lung mechanics and lung fluid balance. J Appl Physiol Respir Environ Exerc Physiol. 1982; 53: 967-976.
- Pauwels RA, Kips JC, Peleman RA, Van Der Straeten ME. The effect of endotoxin inhalation on airway responsiveness and cellular influx in rats. Am Rev Respir Dis. 1990; 141: 540-545.
- Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997; 99: 944-952.
- Song M, Kellum JA. Interleukin-6. Crit Care Med. 2005; 33: S463-465.
- Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999; 282: 54-61.
- Tang WW, Yi ES, Remick DG, Wittwer A, Yin S, Qi M, et al. Intratracheal injection of endotoxin and cytokines. IX. Contribution of CD11a/ICAM-1 to neutrophil emigration. Am J Physiol. 1995; 269: L653-659.