
Review Article
Austin J Endocrinol Diabetes. 2016; 3(4): 1052.
Insulin Resistance Syndrome in OB/OB Mouse
Tatsuo Tomita*
Departments of Integrative Bioscience and Pathology, Oregon Health and Science University, USA
*Corresponding author: Tatsuo Tomita, Department of Integrative Bioscience, Oregon Health and Science University, 611 SW Campus Drive, Portland, USA
Received: August 23, 2016; Accepted: September 23, 2016; Published: October 14, 2016
Abstract
The ob/ob mouse had been extensively used as a good animal model for insulin resistance study especially obesity associated Type 2 diabetes (T2DM) in humans. Ob/ob mice develop obesity as early as 1 month of age with progressively increasing body weight and insulin levels in both plasma and pancreas due to increasing insulin resistance through leptin deficiency in the hypothalamus. Progressively hyperplastic and hypertrophic islets in ob/ob mice consist predominantly of β-cells and correspond to extreme hyperinsulinemia and hyperglycemic ob/ob mice with age responding to progressive insulin resistance with hyperglycemia. Abnormal glucose and insulin tolerance tests revealed increasing higher plasma glucose and insulin levels in ob/ob mice with age and support increasing insulin resistance with age. This study aimed to reveal increasing insulin resistance in ob/ob mice with aging, similar to humans with type 2 diabetes. Leptin treatment and leptin gene therapy dramatically improve body weight with normalizing glucose tolerance and insulin tolerance test. Insulin resistance is partly due to decreased insulin receptor sites in the target organs and ob/ob mice have been used to unfold biological interactions of Insulin-Receptor Substrate -1 (IRS-1) and IRS-2 and may elucidate a complex mechanisms of a kinase cascade, through which insulin resistance will be further analyzed at the molecular level.
Keywords: β-cells; glucose; Hyperphagia; Insulin; Insulin receptor; Insulin resistance; IRS-1; IRS-2; Islets of Langerhans; Leptin; NPY
Abbreviations
ISR: Insulin-Receptor Substrate; NPY: Neuropeptide Y; T2DM: Pi (3) kinase [PI (3) K] and type 2 diabetes
Introduction
Leptin-deficient ob/ob mouse was discovered at the Jackson Memorial Laboratory, Bar Harbor, Maine in 1949 [1]. A non-obese mouse colony was raised and produced a strain of obese offspring, which suggested that a single mutation had occurred in a hormone, leptin, regulating hunger and energy expenditure [1]. Leptin was the first fat cell-derived hormone to be discovered [2] and has the structure of a long chain helical cytokine and is expressed in adipose tissue in proportion to adipocyte size [3,4]. Leptin acts on receptors in the lateral hypothalamus to inhibit hunger by counteracting the effects of Neuropeptide Y (NPY), a potent huger promoter secreted by cells in the gut and in the hypothalamus [2] and also acts on the medial hypothalamus to stimulate satiety [1]. Absence of leptin in ob/ob mice leads to hyperphagia and resulting extreme obesity [2]. Ob/ob mice are indistinguishable from the lean littermates at birth but become heavier within 1 month after birth [5]. Insulin resistance progresses with aging through ever increasing obesity in ob/ob mice, similar to aging humans with type 2 diabetes [2]. Obese human individuals generally exhibit more leptin in the blood than lean individuals due to more leptin secreting fat cells and exogenous leptin injection does not reduce obesity in humans in resistance to leptin [6]. In a contrast, ob/ob mice will revert obesity to normal weight by exogenous leptin injection or leptin gene treatment [6-8].
Pathophysiological study
Ob/ob mice were indistinguishable from lean littermates at birth but already had a sign of chubby appearance with 1/3 heavier body weight within 1 month of age (Figure 1) [5,9]. Plasma glucose levels were already twice as much as that of lean mice (Figure 1) [9]. Body weight progressively increased up to 6 months reaching 70 g as twice as heavy as lean mice (Figure 1) [9] whereas plasma glucose peaked at 3 months and subsequently decreased in 6 months twice as much as that of lean mice (Figure 1) [9]. Glucose tolerance test, 2 mg glucose per g of body weight intraperitoneal, ip, revealed a marked difference of plasma glucose and insulin levels between ob/ob mice and lean mice: lean mice showed consistently high plasma glucose levels at 12—13 mmol/l at 30 min from 1 to 6 months of age whereas 1 month old ob/ob mice revealed the highest plasma glucose of 35 mmol at 60 min, 3.5 fold higher than that of lean mice and both 3 and 6 month old ob/ob mice revealed the highest glucose levels of 30 mmol/l at 30 or 60 min after glucose infusion (Figure 2) [10]. Plasma insulin levels of 1 month old ob/ob mice were about twice as much as that of lean mice and subsequently increased to fivefold that of lean mice at 6 months (Figure 2) [10]. Thus, glucose tolerance test showed much higher plasma glucose and insulin levels in ob/ob mice than in lean mice (Figure 2) [10]. Insulin tolerance test, porcine insulin 150 mU/ml, 1mU/g of body weight, ip revealed that lean mice responded by the lowest plasma glucose to 3 to 4 mmol/l from 30 to 120 minutes after insulin injection whereas 1 month old ob/ob mice revealed the lowest plasma glucose to 7 mmol/l in 1 month of age, 9 mmol/l in 3 months and 10 mmol/l in 6 months old, showing much higher plasma glucose levels, progressively increased from age 1 to 6 months (Figure 3) [10]. Plasma insulin levels were about the same for both ob/ob mice and lean mice of 1 month old (Figure 3) [10]. However, plasma insulin levels of ob/ob mice reached the highest levels at 3 months, peaked at 45 μmol/l of three fold higher than that of lean mice, and slightly decreased until 6 months at 40 μmol/l 30 min after insulin injection (Figure 3) [10]. Plasma insulin levels stayed 3 to 4 fold higher in ob/ob mice than in lean mice (Figure 3) [10]. Pancreatic tissue level of insulin and other pancreatic hormones: Pancreatic insulin content of ob/ob mice was about 30% higher for 1 and 2 month old mice compared to lean mice, but it progressively increased to fivefold at 6 month old while lean mouse insulin content staying at the same levels of 100 μg/g throughout 6 months of age (Figure 4) [9]. Glucagon level was 50% higher for 1 month old ob/ ob mice and also progressively increased to 2.5 fold toward 6 months compared to lean mice (Figure 4) [9]. Pancreatic polypeptide level was twice as that of lean mice at 1 month and moderately higher throughout 6 months (Figure 4) [9]. Thus, in ob/ob mice insulin level progressively increased higher with age of mice, followed by glucagon and PP compared to lean mice but somatostatin level did not increase with age of mice, staying at the same levels of that of lean mice from 1 to 6 months of age (Figure 4) [9]. These normal levels of pancreatic somatostatin correlate with normal levels of serum somatostatin, which will not reduce markedly elevated serum insulin and moderately increased serum glucagon levels [11]. The increasing islet sizes and numbers support ever increasing plasma insulin levels in response to an increasing insulin resistance with age in ob/ob mice [10]. Insulin removal in perfused livers of ob/ob mice: Karakash et al. measured insulin concentration in flow and outflow of perfusate from perfused livers in situ from 2 and 3 months old ob/ob and lean mice: the percentage of the insulin removed by the liver was considerably lower in ob/ob mice than in livers of lean mice using various insulin concentrations of the perfusate [12]. Ob/ob mice were also made normoglycemic by prior injection of streptozotocin or anti-insulin serum: insulin removal was increased by 50% by streptozotocin or anti-insulin serum. These data suggest insulin-binding and removal by the liver were much lower in hyperglycemic and hyperinsulinemic ob/ob mice and removal percentage was increased by normoglycemic ob/ob mice by streptozotocin or anti-insulin antibody injection. Hyperglycemic ob/ob mice are hyperinsulinemic partly due to lesser insulin removal in the liver, the main organ for insulin to bind and be removed. In hyperglycemic ob/ob mice more insulin circulates in the blood as a result of lesser insulin removal in the liver [11,12]. Leptin treatment and leptin gene therapy on ob/ob mice: In 5 week old ob/ob and lean mice, Harris et al. placed an Alzet mini osmotic pump (Alza Corp, Palo Alto, CA) in the peritoneal cavity of each mouse and delivered 0 to 42 μg/day human leptin for 7 days [4]. In ob/ob mice 2 to 42μg/day reduced food intake and body weight [4]. Liver weight, lipid and glycogen decreased 50%, 70% and 47%, respectively [4]. Serum glucose and insulin were reduced in all leptintreated mice and were normalized by 10μg/day leptin [4]. These doses also increased brown adipose tissue as well as increasing rectal temperature and sympathetic nerve activities [4]. Exogenous leptin treatment had shown to successfully revert hyperphagia in ob/ob mice and to reduce body weight through normalizing serum glucose, insulin, liver weight, normal lipid and glycogen content [4]. Ten to 12 months old ob/ob mice were treated with leptin (100μg/o.1 ml), ip for 7 days and their isolated islets secreted improved insulin secretion with 16.7 mmol glucose [13]. In 18 to 22 month old male mice, ob/ ob mice were treated with a recombinant adenovirus expressing the mouse leptin cDNA [8]. Treatment resulted in dramatic reductions in both food intake and body weight as well as normalization in serum insulin levels and glucose tolerance [8]. Subsequent diminishment in serum leptin levels resulted in the rapid resumption of food intake and a gradual body weight gain, correlating with the gradual return of hyperinsulinemia and insulin resistance [8] Most recently, DiSilvestro et al. produced an encapsulated fat pad containing leptin over expressing 3T3-L1 preadipocyte cell line, which was injected into visceral fat deposit in 5 week old ob/ob mice [14]. Leptin was found in the plasma of an encapsulated fat pad injected ob/ob mice but not in the ob/ob mice without fat pad injection at the end of 72 days [14]. Fat pad injected ob/ob mice had transit suppression in appetite and weight loss and had greater brown fat mass, metabolic rate and reduced resist in plasma levels compared to non-fat injected ob/ob mice [14]. By glucose tolerance test, blood glucose levels were significantly improved in fat pad injected ob/ob mice compared to non-fat pad injected ob/ob mice [12]. Thus, an encapsulated fat pad provided a long-term improvement of insulin resistance syndrome in ob/ob mice [14]. In 6 week old mice, gonadal fat pad of white adipose tissue from lean littermates was transplanted subcutaneously in ob/ ob mice, in whom 4 of 8 donors prevented obesity in ob/ob mice despite leptin levels being below 25% of lean littermates [15]. Thirteen month old ob/ob mice were also transplanted subcutaneously with white adipose tissue to test the therapeutic potential, which reduced body weight with reduced non-fasting insulin [15]. In also 6 week old ob/ob mice, brown adipose tissue transplanted from lean littermates reportedly reduced body weight with increased oxygen consumption and decreased fat mass, resulting improved insulin resistance and liver statuses without an evidence of the increased serum leptin levels [15].
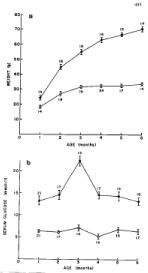
Figure 1: Comparison of body weight and serum glucose levels in ob/ob
and lean mice from 1 to 6 months of age. Plasma glucose levels of ob/ob
mice were already twice as much as lean mice at 1 month of age despite a
moderate body weight gain at that age. Body weight progressively increased
up to 6 months, reaching 70 g as twice as heavy as lean mice whereas
plasma glucose peaked at 3 months and subsequently decreased toward 6
months maintaining plasma glucose twice as much of that of lean mice. •--•:
ob/ob mice, o—o: lean mice, A: Body weight, B: Serum glucose levels [9].
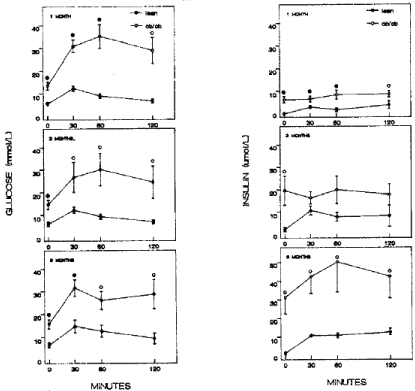
Figure 2: Glucose tolerance test There was a marked difference between ob/ob and meal mice for plasma glucose and insulin levels. Lean mice showed
consistently high plasma glucose levels at 12 to 14 mmol/l at 30 min from 1 to 6 months of age whereas 1 month old ob/ob mice reveled the highest plasma glucose
of 35 mmol/l at 60 min, 3.5 fold higher than that of lean mice and both 3 and 6 month old ob/ob mice revealed highest glucose levels of 30 mmol/l at 30 to 60 min
after glucose infusion (left figure). Plasma insulin levels of 1 month old ob/ob mice were about twice as much as that of lean mice and subsequently increased to
fivefold that of lean mice at 6 months. •--•: ob/ob mice, o—o: lean mice, Left figure: plasma glucose levels, Right figure: plasma insulin levels [10].
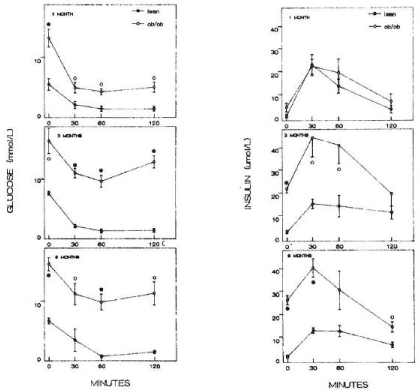
Figure 3: Insulin tolerance test Lean mice responded by the lowest plasma glucose to 3 to 4 mmol/l from 30 to 120 min after insulin injection whereas 1 month old
ob/ob mice showed the lowest plasma glucose of 7 mmol/l in 1 month of age, 9 mmol/l in 3 months and 10 mmol/l in 6 months (left figure). Plasma insulin levels
were about the same for both ob/ob and lean mice of 1 month of age but plasma insulin levels of ob/ob mice reached the highest levels at 3 months, peaked at 45
μmol/l, three fold higher than that of lean mice, and slightly decreased until 6 months at 40 μmol/l. Plasma insulin levels stayed at 3 to 4 times higher in ob/ob mice
than that of lean mice. •--•: ob/ob mice, o—o: lean mice, Left figure: plasma glucose levels, Right figure: plasma insulin levels [10].
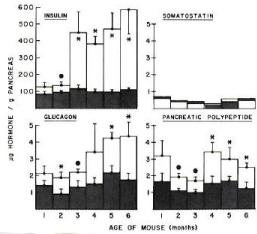
Figure 4: Pancreatic tissue levels of insulin, glucagon, somatostatin and
pancreatic polypeptide Pancreatic insulin content of ob/ob mice was about
30% higher for 1 and 2 month old mice compared to lean mice, but it
progressively increased at fivefold at 6 months of age. Glucagon content was
50% higher for 1 month old ob/ob mice and also progressively increased to
2.5 folds toward 6 months compared to lean mice. Pancreatic polypeptide
content was twice as that of lean mice at 1 month and moderately higher
throughout 6 months. Somatostatin level was about same for both ob/ob and
lean mice from 1 to 6 months of age [9].
Histological and morphometric study of pancreatic islets: Islet area was measured on H. & E. stained sections using HIPAD digital tablet (28 x 28 cm active field, Ram Biometrics, Nashville, TN) in square μm [10]: the total islet area of lean mice was between 0.56 and 0.79 % of the total pancreatic tissue area from 1 to 7 months of age (Figure 5A and E, Table 1) whereas ob/ob mouse islet was more than twice that of lean mice at 1 month of age and progressively increased from fivefold in 2 month to fiftyfold in 7 months of age (Figure 5B,C,D and F, Table 1) [10]. Some hyperplastic islets were located adjacent to the small duct, suggestive of nesidioblastosis or new islet formation from the duct system in 2 month old ob/ob mouse pancreas (Figure 5D). Edema and necrosis started to appear in the midst of extremely large, hyperplastic islets of 4 month to 7 month old ob/ob mice with occasional apoptotic bodies (Figure 5F) [10]. Regarding the mean islet areas, lean mouse islets did not increase drastically with age, staying about the same sizes measuring 10,000 –12,000 μm2 between 1 and 7 months of age whereas ob/ob mouse islets were 20% larger at 1 month of age and progressively increased in 2 to 7 months of age (Table 2) [10]. Not only were there a greater number of islets in ob/ob mouse pancreas but each islet from 6 to 7 months old ob/ob mice was eight to nine times larger than that of lean mice of the corresponding age (Table 2) [10]. Any given sections of the microscopic slide present greater variation of islet sizes in ob/ob mouse islets than lean mouse islets (Figure 5, Table 2) [10]. The pancreas was divided into three lobes including ventral (head portion), dorsal (middle portion) and splenic (tail portion) for regional difference, and more islets were observed in the splenic lobe than the two other lobes in ob/ob mice (Table 1). Pancreatic levels of insulin and glucagon were higher in splenic lobe than other two lobes with insulin levels 90 times more in ob/ob mice than lean mice (Figure 6).
![]()
Age
(mos)Mouse Type
%Islet area
Entire Pancreas
Ventral lobe
Duodenal lobe
Splenic lobe
1
Lean
0.616
0.469
1.041
0.567
oblob
1.489
0.848
1.312
1.939
2
Lean
0.786
0.324
0.612
1.009
oblob
5.296
3.955
3.78
7.051
3
Lean
0.684
0.291
0.319
0.997
oblob
9.334
3.653
14.178
9.55
6
Lean
0.663
0.324
1.026
0.702
oblob
18.424
15.579
18.865
24.616
7
Lean
0.557
0.255
0.536
0.888
oblob
27.609
9.913
21.551
33.506
The total islet area of lean mice was between 0.56 and 0.79% of the total pancreatic tissue area from 1 to 7 months of age. Whereas ob/ob mouse islet was more than twice that of lean mice at 1 month of age and progressively increased from fivefold in 2 months to fiftyfold in 7 months of age [10].
Table 1: Islet area versus total pancreatic tissue.
![]()
Age
(mos)Mouse Type
Islet area(µm)2 (mean±SE)
Total Pancreas
Ventral lobe
Dorsal lobe
Splenic lobe
1
Lean
10,349 ± 1,681
8,183 ± 1,448
12,988 ± 2,686
9,735 ± 956
oblob
12,283 ± 1,810
8,614 ± 1,587
15,028 ± 2, 084
12,903 ± 1,750
2
Lean
9,693 ± 1,406
7,628 ± 853
6,544 ± 1,797
10,048 ± 1,529
oblob
22,031 ± 6,553
25,153 ± 6,344
36,798 ± 9,329
34,936 ± 3,895
3
Lean
12,517 ± 2,218
11,705 ± 2,279
11,245 ± 2,339
11,645 ± 2,048
oblob
26,760 ± 8,819
39,768 ± 11,971
49,608 ± 10,609
35,712 ± 4,180
6
Lean
10,635 ± 2,948
5,804 ± 1,530
16,874 ± 4,740
9,113 ± 2,539
oblob
78,639 ± 2,342
102,596 ± 20,097
71,869 ± 9,991
62,875 ± 7,625
7
Lean
9,995 ± 2,342
8,509 ± 2,403
9,133 ± 2,486
12,238 ± 2,146
oblob
87,217 ± 17,953
37,048 ± 6,131
95,716 ± 26,030
126,533 ± 21,299
Mean lean mouse islets measured 10,000 to 12,000 µm2 between 1 and 7 months of age whereas mean ob/ob mouse islets were slightly larger at 1 month but progressively increased in 2 to 7 months of age. Each islet from 6 to 7 months old ob/ob mice was eight to nine times larger than that of lean mice with a large variation of sizes [10].
Table 2: Mean islet area.
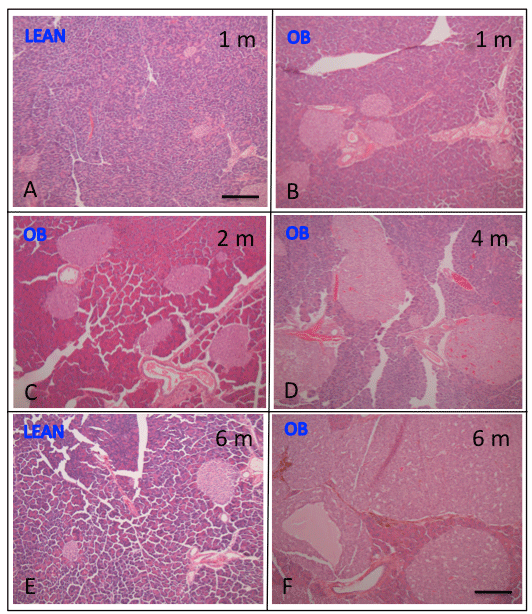
Figure 5: Histology of pancreatic islets of ob/ob and lean mice Islets from 1
to 6 months old lean mice were about the same sizes with slightly larger in
6 month old lean mice (E) compared to 1 month old mice (A). Ob/ob mouse
islets were larger at 1 month of age (B) and progressively increased from 2
(C) to 6 months of age (F) than that of lean mice (E). From 4 to 7 months old
ob/ob mice there was degenerative change of central edema and necrosis in
the middle of extremely large islets containing occasional apoptotic bodies
(F). Bar is 100 μm in length. A: 1 month old lean mouse, B: 1 month old ob/ob
mouse, C: 2 month old ob/ob mouse, D: 4 month ob/ob mouse, D: 6 month
old lean mouse, F: 6 month old ob/ob mouse pancreas.
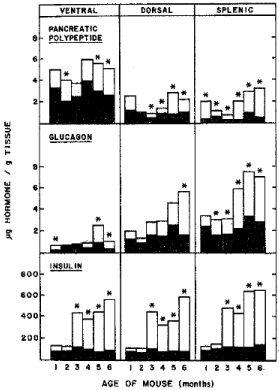
Figure : Pancreatic hormones levels of ob/ob and lean mice by lobes The
pancreas was divided into ventral (head portion), dorsal (middle portion) and
splenic (tail portion). Insulin levels of lean mouse pancreas increased slightly
with age whereas insulin levels of ob/ob mice increased markedly with age at
about six fold insulin levels at 6 months of age compared to that of 1 month
of age. Glucagon levels increased with age both in ob/ob and lean mice with
more than twice glucagon in four to six month old ob/ob mice than that of lean
mice . In ob/ob mouse pancreas, total insulin levels were about 90 times more
than that of glucagon . Pancreatic polypeptide levels were the highest in the
ventral lobe both in ob/ob and lean mice with ob/ob mouse levels nearly twice
as much as that of lean mice [9].
Biochemical study: The leptin receptor is found in many tissues in several alternatively spliced forms raising the possibility that leptin exerts effects on many issues including the hypothalamus [17]. Leptin injection activates Stat3 but no other STAT protein in the hypothalamus of ob/ob and wild type mice but not in db/db mice, mutants that lack an isoform of the leptin receptor [17]. Leptin activates Stat3 was dose dependent [18]. The hypothalamus is a direct target of leptin action and this action is critically dependent on gp-130-like leptin receptor isoform [17]. Insulin-receptor interaction in liver cell membrane from ob/ob mice: Kahn et al initially reported on insulinreceptor interaction in the purified liver cell membrane and isolated hepatocytes from ob/ob and lean mice [17]. Liver cell membranes from ob/ob mice bind only 20-25% as much insulin per mg protein as those of lean mice [16]. Scachard analysis suggests that this decrease in binding is due to a decrease in insulin receptor sites in the liver cell membrane, especially those of higher affinity [18]. This decrease in insulin receptor sites corresponds to the decreased numbers of fat-fat cells from type 2 diabetic subjects with obesity to support that ob/ob mice is a model for human type 2 diabetes with obesity-associated insulin resistance [6]. There is also a decrease in [I-125]-glucagon binding but to a much smaller extent than that observed in insulin binding [16]. Isolated hepatocytes from ob/ob mice is larger than that of lean mice but these hepatocytes bind significantly less to [I-125]- insulin especially at the lower insulin concentration of 0.1 to 1.0 ng/ml than those of lean mice [18]. Insulin resistance is also characterized by decrease in kinase activity, the concentration and phosphorylation of IRS-1 and IRS-2, Pi [3] kinase [PI [3] K] activity, glucose transporter translocation and activity of intracellular enzymes [19,20]. In 8 to 10 week old male ob/ob and lean mice, Kerouz et al studied Insulin Receptor (IRS) expression: IRS-1 and IRS-2 are decreased ~ 50% in muscle whereas in liver the decrease is significantly greater for IRS-1 (72%) than for IRS-2 (29%) [21]. In the ob/ob liver there is a change in expression of the alternately spliced isoforms of regulatory subunits for IRS-1-associated phosphatidylinositol PI 3- kinase [PI [3] K] and is 45% decrease in p85α form of [PI [3] K], a nine-fold increase in AS53/p55α and twofold increase in p50α isoforms [21]. There are multiple alterations in the early step of insulin signaling in the ob/ob mice, with differential regulation of IRS-1 and IRS-2, various PI 3-kinase regulatory isoforms and a lack of compensation for the decrease in insulin signaling by any of the known alternative pathways at these levels [22]. A kinase cascade that mediates the posttranscriptional action of insulin in liver and other tissue has been defined [22]. This cascade begins when insulin binding activates the insulin receptor tyrosine-kinase, which polyphosphorylates several docking proteins, most prominently IRS-1 and IRS-2 [22]. Phosphorylated tyrosine on IRS-1 and IRS-2 form attachment sites for signaling molecules including the 85 kDA regulatory subunit of [PI [3] K] [16]. Chronic hyperinsulinemia in 3 month old ob/ob mice down regulates the mRNA for IRS-2, essential component of the insulin-signaling pathway in liver, thereby producing insulin resistance [23]. Despite IRS-2 deficiency in ob/ob mice, insulin continues to stimulate producing of SREP-1c, a transcription factor that activates fatty acid synthesis [19]. The combination of insulin resistance and elevated lipogenesis establishes a vicious cycle that aggravates hyperinsulinemia and insulin resistance in ob/ob mice [23-26].
Conclusion
Leptin-deficient ob/ob mice had been widely used ever since the discovery of leptin mutations in an animal model for insulin resistant type 2 diabetes in humans [4-6,9]. Leptin suppresses appetite by inhibiting the effects of NPY in the lateral hypothalamus and stimulates satiety factor in the medial hypothalamus [4,6,7]. Leptin deficiency in ob/ob mice leads to hyperphagia with resulting extreme obesity through reduced metabolic rate and body temperature as early as 1 month of age [7]. The improvement of insulin resistance syndrome is dramatic by exogenous leptin injection or leptin gene treatment, leading to an immediate improvement of blood glucose and insulin levels within hours of administration and long-time improvement in glucose tolerance and insulin tolerance test with reducing food intake and body weight [7]. Histological finding of pancreatic islets is striking for islet hypertrophy with early hyperplasia from 1 month to ever increasing islet sizes and numbers in 6 months of age and eventually islets develop degenerative, central necrosis of mid-islet through β-cell apoptosis [10]. Thus, ob/ob mice had already provided enormous amount of basic and clinical information on insulin resistance syndrome from insulin receptor study, regulatory subunit of IRS-1 and IRS-2 to a kinase cascade in the insulin target organs in liver, fat and muscle [12-17]. Leptin may have major effects through direct actions on target organs, including β-cells, liver, fat and muscle [26-28]. Despite an initial hope for possible clinical application of leptin in obesity-associated insulin resistance in humans [23], there are few human cases of leptin or leptin-receptor mutations and these humans do not have major insulin resistance with less incidence of T2DM [29,30]. Obese individuals have ample visceral fat with large cytoplasm, producing and secreting ample leptin into the blood, and do not respond to leptin treatment due to mutations of leptin receptor [7]. Among monogenic obese mouse model, ob/ob (Lep ob/ob) mice is the most widely used model with a mutated leptin and another widely used model is db/db (Lepr db/db) mouse with a mutated leptin receptor [31] and both ob/ob and db/db mice are autosomal recessive [4,5]. Polygenic models of obesity provide a more accurate model for T2DM in humans [32]. The polygenic obese mouse models include NZO mice and KK mice [32]. A relative new model is the Akita mouse, which is a genetically induced, non-obese model of β-cell dysfunction due to ER stress and is autosomal dominant [33,34].
References
- Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950; 41: 317-318.
- Conde J, Scotece M, Gomez R, Lopez V, Gonez-Reino JJ, Lago F, et al. Biofactors adipokines: biofactors from white adipose tissue: A complex hub among inflammation, metabolism and immunity. Biofactors. 2011; 37: 413-420.
- Hamilton BS, Paglia D, Kwan AY, Deital M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat Med. 1995; 1: 953-956.
- Harris RB, Ramay TG, Smith SR, Rbuch RC. Early and late stimulation of ob mRNA expression in meal-fed and overfed rat. J Clin Invet. 1996; 97: 2020-2026.
- Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979; 59: 719-809.
- Lindstrom P. The physiology of obese-hyperglycemic mice [ob/ob mice]. Scient World J. 2007; 7: 666-685.
- Harris RB, Zhou J, Redman SM, Smagin GN, Smith SR, Rodgers E, et al. A leptin dose-response study in obese (+/?) mice. Endocrinol. 1998; 139: 8-19.
- Muzzin P, Eisensmith PC, Copeland KC, Woo SLC. Correction of obesity and diabetes in genetically obese mice by leptin gene therapy. Proc Natl Acad Sci USA. 1996; 93: 14804-14808.
- Tomita T, Doull V, Kimmel J, Pollock HG. Pancreatic polypeptide and other hormones in pancreas of obese (ob/ob) mice. Diabetologia. 1984; 27: 454-459.
- Tomita T, Doull V, Pollock HG, Krizsan D. Pancreatic islets of obese hyperglycemic mice (ob/ob). Pancreas. 1992; 7: 367-375.
- Quesada I, Tudur E, Pipoll C, Nadal A. Physiology of pancreatic α-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008; 199: 5-19.
- Karakash, C, Assimacoupoulous-Jannet F, Jeanrenaus B. An anomaly of insulin removal in perfused livers of obese-hyperglycemic (ob/ob) mice. J Clin Invest. 1976; 57: 1117-1124.
- Khan A, Narangoda S, Ahren B, Holm C, Sundler F, Effendic S. Long-term leptin treatment of ob/ob mice improves glucose-induced insulin secretion. Int J Obesity. 2001; 25: 816-821.
- DiSilvestro DJ, Melgar-Bermudez E, Yasmeen R, Fadda P, Lee Li, Kalyanasudaram A, et al. Leptin production by encapsulated adipocytes increases brown fat, decreases resistin, and improves glucose intolerance in obese mice. PLOS One. 2016; 11: 0153198.
- Klebanov S, Astle CM, DeSimone O, Ablamunits V, Harrison DE. Adipose tissue transplantation protects ob/ob mice from obesity, normalizes insulin sensitivity and restores fertility. J Endocrinol. 2005; 186: 203-211.
- Lie X, Wang S, You Y, Meng M, Zheng Z, Dong M, et al. Brown adipose tissue transplantation reverses obesity in ob/ob mice. Endocrinol. 2015; 156: 2461-2469.
- Vaisse C, Halaas J, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nature Genetics. 1996; 14: 95-97.
- Kahn CD, Neville DM Jr, Roth J. Insulin-receptor interaction in the obese-hyperglycemic mouse. A model of insulin resistance. J Biol Chem. 1973; 248: 244-250.
- Pessin JE, Saitiel AR. Signaling pathways in insulin action: molecular target of insulin resistance. J Clin Invest. 2000; 106: 165-169.
- Saitiel AR, Kahn R. Insulin signaling and regulation of glucose and lipid metabolism. Nature. 2001; 414: 799-806.
- Kerouz N, Horsch D, Pons S, Kahn R. Differential regulation of insulin receptor substrates-1 and 2 (IRS-1 and IRS-2) and phosphatidylinositol 3-kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J Clin Invest. 1997; 100: 3164-3172.
- White MF. The IRS-1 signaling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998; 182: 3-11.
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-1 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000; 6: 77-86.
- Plleymovater MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995; 269: 540-543.
- Larcher F, Del Rio M, Serrano F, Segovia JC, Ramirez A, Meana A, et al. A cutaneous gene therapy approach to human leptin deficiencies correlation of murine ob/ob phenotype using leptin –targeted keratinocyte graft. FASEB J. 2001; 15: 1529-1538.
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000; 106: 473-481.
- Kim YB, Ootani S, Pirroz DD, Flier JS, Kahn BB. In vivo administration of leptin activates signal transduction directly in insulin sensitive tissues: overacting but distinct pathways from insulin. Endocrinol. 2000; 141: 2328-2339.
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chair SL, Rabinowitz O, et al. Weight-reducing effects of plasma protein encoded by the obese gene. Science. 1995; 269: 543-546.
- Montague CT, Farooqi IS, Whitehead JP, Soos MA, au H, Warenham NJ, et al. Congenital leptin deficiency is associated with severe early onset obesity in humans. Nature. 1997; 387: 903-908.
- Clements K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998; 392: 398-401.
- Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996; 84: 491-495.
- King AJF. The use of animal models in diabetes research. Br J Pharmacol. 2012; 166: 877-894.
- Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997; 46: 887-894.
- Makino S, Kunimoto K, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980; 29: 1-13.