
Case Report
Austin J Endocrinol Diabetes. 2017; 4(1): 1054.
21-Hydroxylase Deficiency Presenting as Bilateral Adrenal Masses in a Phenotypically Male but Genetically Female Patient
Evgenia Panagiotidi¹, Georgios Papadakis¹*, Eleni Triantafillou¹, Paraskevi Manitarou¹, Styliani Kalaitzidou¹, Aggeliki Sapera¹, Victoria Kaltzidou¹, Maria Dracopoulou², Gino Veccini³ and Athanasia Tertipi¹
¹Department of Endocrinology, Metaxa Anticancer Hospital, Greece
²Department of Pediatrics, Athens University Medical School, Greece
³Department of Pathology, Metaxa Anticancer Hospital, Greece
*Corresponding author: Georgios Papadakis, Department of Endocrinology, Metaxa Anti-Cancer Hospital, Piraeus, Greece, Botasi 51, 18537, Pireaus, Athens, Greece
Received: December 01, 2016; Accepted: January 25, 2016; Published: February 02, 2017
Abstract
A 59-year-old male was investigated for bilateral adrenal masses. At birth he had a phallus and partial fusion of the labioscrotal folds and was raised as male. At puberty he menstruated and underwent multiple operations for the creation of a scrotum and the restoration of hypospadias. The uterus and ovaries were removed and prosthetic testes were placed in the scrotum. He was not receiving any cortisone supplementation. Genetic testing of the 21-hydroxylase gene revealed a compound heterozygoticity for the mutations p.I172N and p.Q318X. Following left adrenalectomy histology revealed a diffusely hyper plastic adrenal cortical zone with regions of myelolipoma transformation. In patients with Congenital Adrenal Hyperplasia there is an increased frequency of adrenal adenomas and myelolipomas related to the chronic stimulation of the adrenal cortex by ACTH and adrenal androgens. Adrenal myelolipomas are benign and biochemically inactive neoplasms. Measurement of17-hydroxyprogesterone in cases of adrenal enlargement can lead to the diagnosis of Congenital Adrenal Hyperplasia.
Keywords: 21-Hydroxylase deficiency; Congenital adrenal hyperplasia; Adrenal myelolipoma
Introduction
Recently, there have been reported several cases of the classic form of Congenital Adrenal Hyperplasia (CAH) diagnosed in adult life, only after the incidental discovery of adrenal masses on Computed Tomography (CT). Here we report a 59-year-old phenotypically male but genetically female patient with the Simple Virilizing (SV) form of 21-Hydroxylase Deficiency (21-OHD), previously undiagnosed who presented with bilateral adrenal masses, described as myelolipomas on CT.
Methods and Results
A 59 year-old male, followed by the Hematological Department of our Hospital for polycythemia was referred to the Endocrinological Department for the investigation of bilateral adrenal masses found incidentally on abdominal CT. The adrenal masses measured 5 cm on the right and 8 cm on the left adrenal (Figure 1), both with features characteristic for myelolipoma. Family history was not contributory.
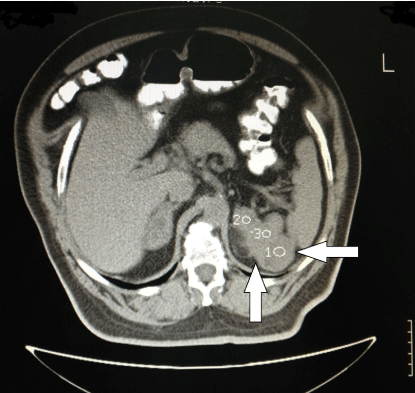
Figure 1: Adrenal CT of the patient. The left adrenal mass is seen. The
Housfield Units are 1-3 with >60% washout at 15’.
The patient was assigned the male gender and raised as a man, since at birth he had a phallus and partial fusion of the labioscrotal folds.He was considered bilaterally cryptorchid and hypospadias.He reported being the taller boy among his classmates initially, but stopped growing at the age of 10. At the age of 15 the patient menstruated and the karyotype testing identified the 46, XX chromosomal sex. The patient at that time strongly refused a gender reassignment and was submitted on multiple operations in another hospital, for the creation of a scrotum and the restoration of hypospadias.The uterus andthe ovaries were removed and prosthetic testes were placed in the scrotum. The patient denied any clinical symptoms suggestive of adrenocorticaldeficiency. To be noticed, most of the details regarding the diagnosis at the age of 15 were reported by the patient’s sister. The patient himself hid his genetic sex and did not accept to undergo any special psychological test. However, he looked quite concordant psychologically with the gender of assignment, he had worked as a cook in the merchant fleet and he even had had an unsuccessful marriage. On physical examination he had a male appearance with sparse beard and male type baldness; his height was 138 cm, weight 73 kg, and body mass index 38.3 Kg/m². He had a phallus with a 5 cm length and a 2.5 cm circumference, a well formed scrotum with very hard, clearly prosthetic testis of 15 ml volume. The urethral orifice was at the top of the phallus. Gynecomastia was also present. Hormonal laboratory dataare summarized in Table 1.
![]()
Hormone
Value
Normal Range for menopausal women
Testosterone(ng/ ml)
7.8
0.07-0.65
DHEAS (μg/dl)
420
22-263
E2 (pg/ml)
144
5.7-102
ACTH (pg/ml)
80.5
7.3-63.3
PRA (ng/ml)
3.6
0.6-1.9
Aldosterone (pg/ ml)
450
66-173
Synachten test
Time
17(OH)P (ng/ml)
Cortisole(nmol/l)
0’
92
(Normal Range 0.19- 0.71)
199
(Normal Range 260-720)
30’
139
(Normal Range <10)
242
(Normal Range >500)
Table 1: Basic hormone levels of the patient.
Karyotype showed 46, XX and genetic analysis of 21-hydroxylase gene revealed that the patient was a compound heterozygote for the mutations p.I172N and p.Q318X (Figure 2). DNA was extracted from peripheral blood leucocytes using standard methods. The CYP21A2 gene was selectively amplified against the highly homologous CYP21A1P pseudogene in three different overlapping PCR fragments. All 10 exons, 9 introns, 420 bp of the 5΄ untranslated region and 200 bp of the 3΄ untranslated region of the CYP21A2 gene were directly sequenced. Sequencing reactions were performed using the Applied Biosystems Big Dye terminator cycle sequencing kit and analysed on an ABI 3500 Genetic Analyzer.
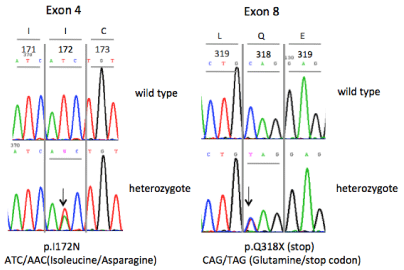
Figure 2: CYP21A2 GENE. The patient was found compound heterozygote
for the mutations p.I172N and p.Q318X of the CYP21A2 gene. The specific
genotype is associated with the classic simple virilizing form of 21-hydroxylase
deficiency (C-SV CAH).
The patient underwent a left adrenalectomy and the histology revealed a huge adrenal measuring in total 12x8x4 cm with diffusely hyperplastic adrenal cortex and regions of myelolipoma transformation (Figure 3).
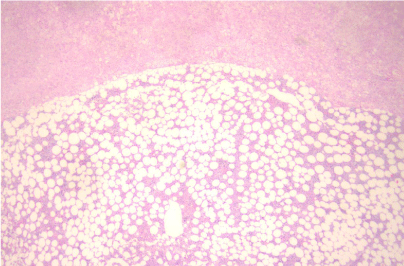
Figure 3: Hematocilin/ Eosin x 250: Hyperplastic adrenal gland cortex in the
upper part of figure with myelolipoma loci in the lower part.
Discussion
CAH is a group of inherited, autosomal, recessive disorders characterized by impaired synthesis of cortisol and, in most instances, increased synthesis of adrenal androgens. The defect lays on mutations in several enzymes of the steroid pathway and its prevalence ranges from 1:10,000 to 1:20,000 births [1]. 21-OHD is by far the most common among the various enzymatic deficiencies, accounting for 90-95% of CAH cases, leading to decreased production of cortisol and aldosterone [2]. There are 3 forms of 21-OHD, distinguished by the clinical expression of the disease: 1) the Salt-Wasting form (SW) that presents with adrenal crisis in the first weeks of life and virilization of the female newborn, 2) the Simple Virilizing form (SV), where there is only virilization of the external genitalia in the female infant, and 3) the milder, without any of the above features, Non Classical form (NC) [3]. The two classic phenotypes of CAH, the SW and SV forms, are diagnosed usually in the first weeks of life due to the adrenal crisis ensuing in the SW form, in addition to ambiguous external genitalia in the female newborn and the presence of ambiguous genitalia only in the SV form. The NC form is diagnosed during puberty due to early adrenarche or in early adult life with signs and symptoms of hyperandrogenism in women. All three forms are caused by mutations of the CYP21A2 gene encoding 21-hydroxylase which converts 17-hydroxyprogesterone to 11-deoxycortisol and progesterone to deoxycorticosterone, the respective precursors for cortisol and aldosterone.
More than 100 mutations of the CYP21A2 gene have been identified [4]. It is located on the short arm of chromosome 6, within the region of the major histocompatibility complex, at a short distance from a highly homologous (>95%) pseudogene, designated CYP21A1P [5]. Despite its high homology with CYP21A2, CYP21A1P is totally inactive. p.Q318Xmutation in exon 8 causes complete loss of the enzymatic activity of 21-OH and is present in CYP21A1P. Thus, the mutation is thought to be created by gene conversion between the CYP21A1P and the CYP21A2 during meiosis. In the Greek population it is present in 14.3% of patients with the classic form, either SW or SV [6]. On the contrary, the p.I172N mutation in exon 4 leads to an enzymatic activity of around 2% of the wild-type CYP21A2 gene [7] and it is found in the SW and the SV forms of classic 21-OH deficiency. In the Greek population it is present in 35.3% of patients with the SV form [6]. Therefore, our patient harbored two relatively common for the Greek population mutations of the CYP21A2 gene, causing the classic form of CAH and since the phenotype is determined by the least harmful mutation, the clinical picture was that of the SV form.
Adrenal myelolipomas are rare, benign and biochemically inactive neoplasms, of varied composition of adipose and hemopoietictissue. They frequently are diagnosed as adrenal incidentalomas, as they are usually asymptomatic, although they can grow very large and cause pressure symptoms to the surrounding structures, or even hemorrhage [8,9]. Diseases associated with increased ACTH, such as Cushing’s disease, Addison disease and CAH have been associated with the development of myelolipomas [10]. Till now, no more than 40 cases of CAH associated with myelolipomas[10,11,12] have been described, frequently with very large size [11,13,14]. The patients may or may not been diagnosed with CAH prior to the detection of the adrenal masses. Bilateral or unilateral adrenalectomy is the treatment of choice.
There are three interesting issues in our case. Firstly, the absence of symptoms or signs of adrenal deficiency, despite the inadequate response of cortisol to Synacthen test, compatible with cortisol deficiency. It is conceivable that the minimal enzymatic activity of 21-OH allowed by the p.I172N mutation may have prevented the development of adrenal deficiency. Patients with the SV form maintain an adequate aldosterone production and normal sodium balance at the cost of increased plasma renin activity [15], as was the case in our patient. The enormous mass of the adrenal cortex may account for the increased levels of serum aldosterone.
Secondly, the development of gross enlargement of the adrenal cortex secondary to the minimal, yet long standing, elevation of ACTH. In patients with CAH, especially in those sub optimally treated, there is an increased relative frequency of adrenal adenomas and myelolipomas [16]. The most obvious cause would be the chronic stimulation of the adrenal cortex by ACTH and the adrenal androgens. Efforts to prove such a hypothesis have shown conflicting results, since some tumors from patients with CAH show increased expression of the ACTH and the androgen receptors, while others are not [17,18]. Therefore, the pathogenetic mechanisms involved in the formation of myelolipomas remain obscure. Nevertheless, it is prudent to measure 170HP in cases with adrenal enlargement, not only to obtain the correct diagnosis [19] but also to identify the very rare, still existing, patients with CAH who may harbor an adrenal carcinoma [20].
A third remarkable point in our case is the seemingly at least, absolute concordance of the patient’s life. He chose a profession stereotypically more practiced by men and a female sexual partner. It is reported that patients like ours, diagnosed during puberty or as adults and reared as males, show male psychological and heterosexual orientation [8]. This case supports the option of choosing the male gender of rearing in severely virilized 46, XX infants with CAH.
Statement of Ethics
Subject has given his informed written consent and the study protocol was approved by an appropriate ethics committee.
References
- Van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. European journal of endocrinology / European Federation of Endocrine Societies. 2004; 151: 71-75.
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine reviews. 2000; 21: 245-291.
- White PC, New MI. Genetic basis of endocrine disease 2: congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The Journal of clinical endocrinology and metabolism. 1992; 74: 6-11.
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2010; 95: 4133-4160.
- Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proceedings of the National Academy of Sciences of the United States of America. 1986; 83: 2841-2845.
- Dracopoulou-Vabouli M, Maniati-Christidi M, Dacou-Voutetakis C. The spectrum of molecular defects of the CYP21 gene in the Hellenic population: variable concordance between genotype and phenotype in the different forms of congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism. 2001; 86: 2845-2848.
- New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 2611-2616.
- Wani NA, Kosar T, Rawa IA, Qayum A. Giant adrenal myelolipoma: Incidentaloma with a rare incidental association. Urology annals. 2010; 2: 130-133.
- Kenney PJ, Wagner BJ, Rao P, Heffess CS. Myelolipoma: CT and pathologic features. Radiology. 1998; 208: 87-95.
- German-Mena E, Zibari GB, Levine SN. Adrenal myelolipomas in patients with congenital adrenal hyperplasia: review of the literature and a case report. Endocrine practice: official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2011; 17: 441- 447.
- Ioannidis O, Papaemmanouil S, Chatzopoulos S, Paraskevas G, Konstantara A, Kotronis A, et al. Giant bilateral symptomatic adrenal myelolipomas associated with congenital adrenal hyperplasia. Pathology oncology research: POR. 2011; 17: 775-778.
- Ferreira F, Martins JM, do Vale S, Esteves R, Nunes G, Carmo I. Rare and severe complications of congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a case report. Journal of medical case reports. 2013; 7: 39.
- McGeoch SC, Olson S, Krukowski ZH, Bevan JS. Giant bilateral myelolipomas in a man with congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism. 2012; 97: 343-344.
- Kale G, Pelley EM, Davis DB. Giant myelolipomas and inadvertent bilateral adrenalectomy in classic congenital adrenal hyperplasia. Endocrinology, diabetes & metabolism case reports. 2015; 150079.
- Forest MG. Recent advances in the diagnosis and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Human reproduction update. 2004; 10: 469-485.
- Oliva A, Duarte B, Hammadeh R, Ghosh L, Baker RJ. Myelolipoma and endocrine dysfunction. Surgery. 1988; 103: 711-715.
- Hagiwara H, Usui T, Kimura T, Tagami T, Naruse M, Minamiguchi S, et al. Lack of ACTH and androgen receptor expression in a giant adrenal myelolipoma associated with 21-hydroxylase deficiency. Endocrine pathology. 2008; 19: 122-127.
- Almeida MQ, Kaupert LC, Brito LP, Lerario AM, Mariani BM, Ribeiro M, et al. Increased expression of ACTH (MC2R) and androgen (AR) receptors in giant bilateral myelolipomas from patients with congenital adrenal hyperplasia. BMC endocrine disorders. 2014; 14: 42.
- Nermoen I, Rorvik J, Holmedal SH, Hykkerud DL, Fougner KJ, Svartberg J, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clinical endocrinology. 2011; 75: 753-759.
- Hayashi M, Kataoka Y, Sugimura Y, Kato F, Fukami M, Ogata T, et al. A 68-year-old phenotypically male patient with 21-hydroxylase deficiency and concomitant adrenocortical neoplasm producing testosterone and cortisol. The Tohoku journal of experimental medicine. 2013; 231: 75-84.