
Review Article
J Endocr Disord. 2016; 3(1): 1022.
New Insights on Diabetic Nephropathy
Dahan I, Farber E, Jabali H and Nakhoul F*
Nephrology and Hypertension Division, Bar-Ilan University, Israel
*Corresponding author: Nakhoul F, Nephrology and Hypertension Division, Baruch-Padeh Poriya Medical Center Faculty of Medicine in Galilee Bar Ilan University, Ramat Gan, Israel
Received: April 28, 2016; Accepted: July 14, 2016; Published: July 18, 2016
Abstract
Diabetic Nephropathy (DN) is a long-standing complication of Diabetes Mellitus (DM) and is responsible for more than 40% of end-stage renal disease cases in developed countries. The pathogenesis of DN is multifactorial inclusing genetic and environmental factors. Traditional risk factors and glycemic control are important but inadequate for predicting the incidence and severity of DN. Different pathways are involved in the pathogenesis of DN. Hyperglycemia accelerates oxidative stress with increased production of free radicals.
Reactive oxygen species, particularly those derived from iron, have been implicated in the increase of oxidative stress injury in the Proximal Convolute Tubules and glomeruli with progression of DN. Polymorphic genetic loci encoding variants in enzymes protecting against iron-induced oxidative stress and apoptosis, serve as potential susceptibility determinants for the development of DN. The major function of the Haptoglobin protein is to bind and modulate the fate of extra-corpuscular hemoglobin and its iron cargo. Since Iron plays a major role in the development of DN it may be a therapeutic target for slowing the nephropathy progression.
A combination of glycemic and blood pressure control, utilizing renin angiotensin-aldosterone system RAAS blockers, has been a mainstay of treatment to slow DN progression. Inhibition of RAAS plays a pivotal role in treatment of chronic diabetic kidney diseases. However, reversal of the course of DN or at least long-term stabilization of renal function is often difficult to achieve, and many patients still progress to end-stage renal disease.
In the current review we suggested some integrated mechanisms involved in DN progression.
Keywords: Diabetic nephropathy; Oxidative stress; Klotho; Autophagy; Vitamin- D
Abbreviations
ACEI: Angiotensin Converting Enzyme Inhibitor; ATG: Autophagy-Related Genes; CDKD: Chronic Diabetic Kidney Disease; DN: Diabetic Nephropathy; DM: Diabetes Mellitus; VDR: Vitamin D Receptor; ER: Endoplasmic Reticulum; FGFR1: Fibroblastic Growth Factor-Receptor 1; Hp: Haptoglobin; HG: Hyperglycemia; PCT: Proximal Convolute Tubule; RAAS: Renin Angiotensin Aldosterone System; ROS: Reactive Oxygen Species; SNP’s: Small Nucleotide Polymorphism; TGF- β: Transforming Growth Factor β
Introduction
Diabetic Nephropathy (DN) is the leading cause of end-stage renal disease and dialysis throughout the world. The establishment of novel, effective therapeutic strategies is, therefore required. Proteinuria and/or albuminuria are a sign of glomerular lesions in DN. These lesions can subsequently develop into tubulointerstitial lesions that lead to renal dysfunction. Clinically, therefore, reducing proteinuria is considered a principal therapeutic target to improve renal outcomes in patients with DN. The pathogenesis of DN involves altered intracellular metabolism associated with hyperglycemia, including the activation of protein kinase C, the accumulation of advanced glycation end-products, oxidative stress, altered apoptosis and autophagy [1]. Moreover, hemodynamic changes such as systemic and glomerular hypertension related to hyperactivation of the renin-angiotensin aldosterone system are also involved in DN [2].
Compared with several proteinuric kidney diseases, the renal prognosis of patients with DN is extremely poor. This suggests that the diabetic condition makes various renal cells vulnerable to damage. Cells have evolved several mechanisms to cope with stress and to maintain cellular homeostasis, such as the anti-oxidative stress response and the Endoplasmic Reticulum (ER) stress response [3,4]. In addition, autophagy is an intracellular catabolic processes, in which proteins and organelles are degraded via lysosomes to maintain intracellular homeostasis under certain cytotoxic stress conditions, including hypoxia and ER stress as appears in DN [5]. The recent pathways involved in the generation and progression of DN are the Haptoglobin (Hp) and Klotho proteins, Vitamin D-Vitamin D Receptor and autophagy [6,7].
Haptoglobin and Oxidative Stress
The Haptoglobin (Hp) genotype is a major determinant of progression of nephropathy in individuals with Diabetes Mellitus (DM). We have previously demonstrated an interaction between the Hp genotype and the DM on the accumulation of iron in renal proximal tubule cells [3,4,8]. Transmission electron microscopy demonstrated a marked accumulation of electron-dense deposits in the lysosomes of proximal tubules cells in Hp 2-2 DM mice [3]. These deposits were iron rich, and are associated with lysosomal membrane lipid peroxidation and loss of lysosomal membrane integrity. Cytosolic and mitochondrial Reactive Oxygen Species (ROS) generations were increased notably by Hyperglycemia (HG) treatment. An increase in apoptosis was also observed in cells subjected to HG, which was assessed by the terminal deoxynucleotidyl Transferase- Mediated Digoxigenin Deoxyuridine Nick End-Labeling (TUNEL) procedure. The apoptosis was accompanied with upregulation of Bax and Cytochrome C. Quantitative analyses confirmed that HG induced mitochondrial fragmentation in a time-dependent manner, concurring with increased intracellular ROS production and apoptosis after HG treatment. Autophagy was marginally increased in early treatment with HG [9].
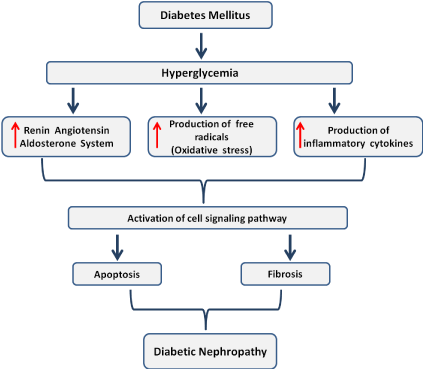
Figure 1: Proposed integrated mechanisms involved in diabetic nephropathy.
Klotho
Klotho is a novel anti-aging gene encoding a protein with a multiple pleiotropic effects. ? Klotho gene is composed of five exons, in humans and mice, is highly expressed in the distal and Proximal Convolute Tubular (PCT) epithelium of normal kidneys. Klotho protein exists in two forms: A membrane and secreted form. The soluble klotho is further well known as anti-apoptotic activity, especially in DN. It’s well known that the expression of klotho in the PCT is decreased in early stages of DN in humans and mice model [7].
Transmembrane klotho is a cofactor that converts Fibroblastic Growth Factor-Receptor 1 (FGFR1) into a specific receptor for FGF- 23 and decreases 1,25(OH)2D3 synthesis in the kidney. 1,25(OH)2D3 stimulates both klotho and FGF-23, and both FGF-23 and klotho inhibits 1,25(OH)2D3 via 1α hydroxylase. It has also been reported that the VDR controls expression of the klotho gene.
Expression of klotho protein is reduced in the kidneys of patients with early DN and klotho gene deficiency exacerbates glomerular injury in diabetic models such as streptozotocin-induced or db/db mice. Up regulation or restoration of klotho by 1,25(OH)2D3, may provide a means to slow down the progression of Chronic Diabetic Kidney Disease (CDKD) and improve cardiovascular disease especially in the DM patients. In addition to the effect of klotho on Transforming Growth Factor β (TGF-β), suppressive effects of klotho on the insulin-like growth factor pathway may be associated with inhibitory action on renal fibrosis and cardio-renal protection in high oxidative stress condition such as diabetes and its complications. We have shown that diabetic Hp 2-2 mice and humans has reduced expression of klotho in the proximal convolute tubules (Unpublished data).
Vitamin D/VDR
Vitamin D is biologically inactive and requires enzymatic conversion to active metabolites. Vitamin D is transported to the liver, where it is first hydroxylated in the 25 position to yield 25-hydroxyvitamin D. Then 25-Hydroxyvitamin D is further hydroxylated by 1-α-hydroxylase in the PCT of the kidney, to yield its active form 1,25-dihyroxyvitamin D. The active 1,25-dihydroxyvitamin D binds to the intracellular Vitamin D Receptor (VDR) to activate vitamin D response elements within target genes. In the kidney, vitamin D is important for maintaining podocytes health, preventing epithelial to mesenchymal transformation and suppressing rennin gene expression and inflammation. 1,25-dihydroxyvitamin D progressively decrease due to PCT injury, leading to a vitamin D deficient state. Additionally, there are some evidences that Vitamin D analogs supplementation (as calcitriol or Paricalcitol) improves the renal damage of diabetic kidney disease patients and their survival [10,11].
Increasing prevalence of diabetes has made the need for effective treatment of DN critical and thereby identifying new therapeutic targets to improve clinical management. Recently Wang et al. showed that vitamin D/VDR signaling in podocytes plays a critical role in the kidney protection from diabetic injury and reconstitution of VDR null mice with the human VDR (hVDR) transgene in podocytes rescued the severe diabetes-related renal damage.
Experimental studies show that administration of a vitamin D analogue reduces albuminuria. Furthermore, 1,25-dihydroxyvitamin D suppressed high-glucose– induced apoptosis of the podocytes in vitro.
Limited data from clinical trials in people with non-diabetic chronic kidney disease suggest that therapy with the vitamin D analogue paricalcitol may reduce proteinuria Furthermore, recent data suggest that paricalcitol, added to Renin Angiotensin Aldosterone System (RAAS) blockade, further reduces albuminuria in people with Type 2 diabetes and diabetic nephropathy.
Although combination therapy showed no additional effect on oxidative system, renin-angiotensin system and renal histology, aliskiren plus paricalcitol significantly decreased interstitial fibrosis volume when compared to monotherapy [12].
Autophagy
The accumulation of damaged proteins and organelles is associated with the pathogenesis of DN. Autophagy, “The self-eating” pathway in the kidney, is activated under some stress conditions, such as oxidative stress and increased free radicals in PCT and in podocytes.
These and other accumulating findings have led to a hypothesis that autophagy is involved in the pathogenesis of DN. Autophagy is a process by which cells degrade and recycle toxic macromolecules and organelles. Emerging body of evidence suggests that targeting the autophagic pathway to activate and restore autophagy activity may be reno-protective [5].
Autophagy is the basic catabolic mechanism that involves cell degradation of unnecessary or dysfunctional intracellular components through lysosomal-dependent mechanism. There are 32 Autophagy-Related Genes (ATG) that are essential for the execution of autophagy. ATG5 is one of the most important ATGs in the autophagy mechanism and it is essential for autophagosome formation and assists in autophagosomal elongation. Recently it was indicated that autophagy plays important roles in many biological processes and diseases such as DN. We are performing new study in diabetic subjects and mice, to evaluate if Specific small Nucleotide Polymorphism (SNP’s) in the ATG5 gene is associated also with DN.
The autophagy-lysosomal degradation pathway is likely to play an essential role in maintaining podocyte function. Podocytes exhibit active autophagy even under non-stress conditions, suggesting that podocytes require a high basal level of autophagy to maintain cellular homeostasis. Podocyte-specific autophagy-deficient mice, resulting from Atg5 gene deletion, have glomerular lesions accompanied by podocyte loss and albuminuria. Furthermore, the impairment of lysosomal function in podocytes by deletion of the mammalian Target of Rapamycin (mTOR), prorenin receptor or mVps34 genes caused severe glomerular sclerosis, massive proteinuria. Since autophagy involves degradation by lysosomes, autophagosomal degradation was disturbed in the podocytes of these mouse models. These results support the idea that the autophagy-lysosomal degradation pathway plays an essential role in maintaining podocyte cell homeostasis.
The physiological role of autophagy in PCT differs from its role in podocytes. Autophagy activity is very low in PCT under basal conditions, but higher rates of autophagy are required by cells under stress conditions. Recent animal studies have shown that autophagy in PCT cells is enhanced during acute kidney injury caused by ischemicreperfusion. Furthermore, mice lacking autophagy activity in PCT, generated by deleting the Atg5 and Atg7 genes, showed progressive renal damage, suggesting that activation of autophagy during acute kidney injury is renoprotective. These mice has a low level of basal autophagy essential to keep cell homeostasis in PCTcells [13].
References
- Mora-Fern´andez C, Dom´inguez-Pimentel V, Mercedes Muros de Fuentes M, G´orriz JL, Mart´inez-Castelao A, Navarro-Gonz´alez JF. Diabetic kidney disease: from physiology to therapeutics. Physiol. 2014; 592: 3997-4012.
- Mauer M, Zinman B, Gardiner R, Drummond KN, Suissa S, Donnelly SM, et al. ACE-I and ARBs in early diabetic nephropathy. J Renin Angiotensin Aldosterone Syst. 2002; 3: 262-269.
- Asleh R, Nakhoul F, Miller-Lotan R, Awad H, Farbstein D, Levy NS, et al. Poor lysosomal membrane integrity in proximal tubule cells of haptoglobin 2-2 genotype mice with diabetes mellitus. Free Radic Biol Med. 15; 53: 779-786.
- Levy AP, Asleh R, Blum S, Levy NS, Miller-Lotan R, Kalet-Litman S, et al. Haptoglobin: basic and clinical aspects. Antioxid Redox Signal. 2010; 12: 293-304.
- Ding Y, Choi ME. Autophagy in diabetic nephropathy. Journal of Endocrinology. 2015; 224: 15-30.
- Kume S, Yamahara K, Yasuda M, Maegawa H, Koya D. Autophagy: Emerging Therapeutic Target for Diabetic Nephropathy. Semin Nephro. 2014; l34: 9-16.
- Guh JY, Chuang LY. Klotho attenuates high glucose-induced fibronectin and cell hypertrophy via the ERK1/2-p38 kinase signaling pathway in renal interstitial fibroblasts. Molecular and Cellular Endocrinology. 2014; 390: 45– 53.
- Nakhoul F, Nakhoul N, Asleh R, Miller-Lotan R, Levy AP. Is the Hp 2-2 diabetic mouse model a good model to study diabetic nephropathy? Diabetes Res Clin Pract. 2013; 100: 289-297.
- Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. 2015; 26: 1304-1321.
- De Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010; 376: 1543-1551.
- Deb DK, Sun T, Wong KE, Zhang Z, Ning G, Zhang Y, et al. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney International. 2010; 77: 1000–1009.
- Erena Mehmet Z, Gunalb Elif Y, Ari Bakirg A, Cobanc J. Caglayanb, Ekimcib N, et al. Effects of Paricalcitol and Aliskiren Combination Therapy on Experimental Diabetic Nephropathy Model in Rats. Kidney Blood Press Res. 2014; 39: 581-590.
- De Rechter S, Decuypere JP, Ivanova E, Van den Heuvel LP, De Smedt H, Levtchenko E, et al. Autophagy in renal diseases. Pediatr Nephrol. 2016; 31: 737–752.