
Special Article - Liver Cirrhosis
Austin J Gastroenterol. 2017; 4(3): 1085.
Sinusoidal Hypertension: A Cause of Liver Cirrhosis in Developing Countries
Shrestha SM*
Liver Foundation, Nepal
*Corresponding author: Santosh Man Shrestha, Liver Foundation Nepal, Kathmandu, Nepal
Received: May 29, 2017; Accepted: June 13, 2017; Published: June 29, 2017
Abstract
Three primary diseases of the liver that cause hepatic venous outflow obstruction (HVOO) are recognized: sinusoidal obstruction syndrome (SOS), Budd-Chiari syndrome (BCS) and hepatic vena cava syndrome (HVCS). Liver cirrhosis develops in these diseases. This paper discusses the pathogenesis of cirrhosis in HVOO.
HVOO results in sinusoidal hypertension. Because of the unique nature of hepatic sinusoid, sinusoidal hypertension is followed by rapid passage of fluid and macromolecules from sinusoid into the space of the Disse and reflex reduction of hepatic arterial flow. As the space between the endothelial cells widens egression of RBCs follows. Hemorrhage and ischemia result in atrophy and apoptosis of hepatocytes in the congested region around the terminal hepatic vein. Extinction of hepatocytes is followed by fibrosis which ultimately leads to development of venocentric cirrhosis. Sinusoidal hypertension also causes reversal of blood flow in portal vein. In BCS and HVCS thrombosis/ thrombophlebitis develops in portal vein and its branches. Atrophy and extinction of hepatocytes supplied by the obstructed portal vein branches lead to development of veno-portal type of cirrhosis in some.
HVCS is endemic in some Asian and African countries. It is a disease related to poor hygienic living condition and caused by bacterial infection. HVOO may develop at anytime in the course disease precipitated by bacterial infection. It is followed by rapid development of ascites and cirrhosis. HVCS induced cirrhosis is characterized by distinctive clinical, histological and ultrasonographic/color Doppler features. HVCS may be a common cause of cirrhosis in developing countries.
Keywords: Sinusoidal hypertension; Liver cirrhosis; Hepatocellular carcinoma
Introduction
Cirrhosis is defined as the presence of fibrous septa throughout the liver that subdivide the hepatic parenchyma into hepatocellular nodules [1]. Cirrhosis results from parenchymal extinction usually caused by obliteration of small portal and hepatic veins. Fibrosis that is formed during acute stage is rapidly removed leaving collapsed preformed stroma. Regenerative activity of the surviving liver cells leads to development of hepatic nodules. The architectural distortion of the liver results in derangement of the blood supply to the hepatic parenchyma. Saline perfusion studies done in hepatic artery showed that whereas in normal liver most of the fluid escaped through hepatic vein, in cirrhotic liver it escaped largely through the portal vein with concomitant rise in portal venous pressure [2]. Similar studies done by perfusing portal vein showed that in normal liver all fluid was recovered from the hepatic vein; but in cirrhotic liver most of it escaped through the collaterals [3]. The perfusion and reconstruction of hepatic vasculature studies in cirrhotic liver also showed presence of anastomotic connections between the hepatic arteries and portal veins that caused transmission of hepatic arterial pressure into portal vein resulting in portal hypertension. Increase portal pressure was further aggravated by resistance to flow caused by narrowing and obliteration of smaller vessels by regenerating nodules [4]. Cirrhosis is thus associated with impaired blood supply to the hepatic parenchyma, development of portal hypertension and opportunity for abnormal growth of liver nodule. Clinical manifestation of cirrhosis is determined by three vectors (a) impairment of parenchymal function, (b) portal hypertension and (c) and features of disease causing cirrhosis, for example that of alcoholic liver disease.
Cirrhosis is a common chronic liver disease throughout the world. Common causes of cirrhosis in different countries vary and it changes in the same country over the course of time. Prevalence of the common cause/s of cirrhosis is determined by geo-cultural factors such as community practice of tattooing, use of alcohol, prevalence of drug abuse and parenteral treatment, living standard and hygiene. In Japan social upheaval that followed the Second World War in late 1940s saw the emergence of intravenous drug abuse that lead to prevalence of hepatitis C infection, which by 1980s had emerged as the major cause of cirrhosis and hepatocellular carcinoma (HCC) replacing alcohol and hepatitis B [5,6]. Similar change in social behavior among youth in 1960-1970s during Vietnam War led to prevalence of hepatitis C through drug abuse that led to its emergence three decades later as an important cause of cirrhosis in USA. The phenomenon was repeated in former Soviet Union after the Afghanistan war [6]. During 1950s veno-occlusive disease due to use pyrrolizidine alkaloid as herbal tea and medication was the common cause of cirrhosis in the West Indies [7]. A peculiar form of cirrhosis called Indian childhood cirrhosis prevailed in India from 1950 to 1990 [8]. In Afro- Asian countries with moderate to high prevalence of chronic hepatitis B, it along with alcohol is regarded as major causes of liver cirrhosis.
Hepatic vena cava syndrome as a common cause of liver cirrhosis
Study done in 1994 showed that prevalence of HBsAg and HCV-RNA among Nepalese patients with cirrhosis was 39% and 8% respectively, and the cause of cirrhosis in Nepal was unknown in approximately 54% of the patients [9]. Further study was done in 2007 [10] to evaluate the role of different known etiological factors of cirrhosis. HBV-DNA was detected in 47%, HCV-RNA in 7% and history of significant alcohol intake (40 g/day in female and 80g/ day in male for 10 years) was obtained in 26% and obstruction of hepatic portion of inferior vena cava (IVC) was detected in 77%. IVC obstruction was a co-morbid condition of patients with chronic hepatitis B and C and alcoholic liver disease. The disease of IVC, later renamed hepatic vena cava syndrome (HVCS) was found to be endemic in Nepal [11]. Long term follow-up of patients showed that 78.5% of the adult patient [12] and 27.5% of children [13] developed cirrhosis. HVCS was thus found to be an important cause of cirrhosis in Nepal.
Hepatic vena cava syndrome (HVCS) is an obliterative disease of the hepatic portion of inferior vena cava, previously known as membranous obstruction of inferior vena cava and was then included under Budd-Chiari syndrome [14]. It was reported from many countries of Asia and Africa [11,15-29]. High incidence of cirrhosis and hepatocellular carcinoma (HCC) was reported in the disease from Japan [15-19] and South Africa [27,28]. HVCS was previously considered a congenital vascular malformation, and diagnosed late in persons with complete obstruction of IVC by cavogram or liver biopsy or detected at autopsy. It is now recognized as a disease related poor hygienic living condition [11,30] and induced by bacterial infection [31]. It usually develops in childhood or young age. A new concept of pathogenesis of the disease has been described recently [32]. Inferior vena cava at the site of opening of the HVs is a junction between upper segment fixed in diaphragm and liver and distal free segment which is prone to micro-endothelial damage and infection. Some persons with bacterial infection develop localized thrombophlebitis at this site which on resolution converts into stenosis or complete obstruction. The circulatory equilibrium is maintained by development of several pathways of cava-caval collateral anastomosis [33]. The obliterative lesion and the collaterals persists the rest of the life. Persons remain asymptomatic for variable periods. Some chronic patients develop hepatomegaly and/or splenomegaly. Persons with splenomegaly may develop features of hypersplenism with thrombocytopenia, leucopenia and anemia.
Acute exacerbations
The abnormal segment however becomes vulnerable to subsequent bacterial infection with deposition of fresh thrombus. The thrombus so formed extends into hepatic veins because of the close proximity of the lesion resulting in acute exacerbations (AE). Minor episodes of AE are recognized by mild jaundice and/or elevation of ALT/AST. Large thrombophlebitis formed during acute stage or severe AE that cause hepatic venous outflow obstruction (HVOO) results in sudden development of ascites from sinusoidal hypertension. About 10% patients develop pleural effusion [34]. Ascites has high protein and high serum albumin ascitic fluid gradient and shows presence of RBC and is invariably associated with mono-microbial bacterial peritonitis due to aerobic organisms like E.coli, Klebsiella or Staphylococcus aureus [35]. Patient with ascites In HVCS has neutrophil leukocytosis, high level of C reactive protein and or bacteremia and has elevated levels of serum bilirubin and ALT/AST. Severe AE may occur at any time in the course of the disease. It commonly occurs in persons with poor nutrition and /or history of alcohol abuse precipitated by chronic diarrhea, puerperal sepsis, surgery or endoscopic procedures [36,37]. Ultrasonography and color Doppler examination of inferior vena cava and liver (Figure 1) is a sensitive and specific method of diagnosis of HVCS [38]. When used with appropriate blood and ascitic fluid tests it also detects minor and severe acute exacerbations.
HVOO is followed by development of cirrhosis from sinusoidal hypertension [12]. Besides HVCS two other primary disease of liver that causes HVOO are recognized (Table 1) where cirrhosis develops from sinusoidal hypertension (Table 1). They are sinusoidal obstruction syndrome (SOS), previously called veno-occlusive disease caused by toxic damage of the sinusoids by pyrrolizidine alkaloids (PA) or myeloablative therapy [39], and Budd-Chiari syndrome (BCS) related to pro thrombotic conditions [40]. This paper describes the prevalence of cirrhosis in the disease of HVOO and discus its pathogenesis, and mentions the distinctive features of cirrhosis due to HBVS, the common cause of HVOO.
![]()
Diseases
1. Sinusoidal obstruction syndrome
2. Budd-Chiari Syndrome
3. Hepatic vena cava syndrome
4. Constrictive pericarditis & congestive heart failure
Table 1: Diseases causing Cirrhosis from Sinusoidal Hypertension.
Diseases of hepatic venous outflow obstruction
Liver is a highly vascular organ that receives blood from two sources. Nearly 80% of the blood entering liver is venous blood rich in nutrients and hormones supplied by portal vein and the reminder well oxygenated blood is supplied by hepatic artery. Blood from these two sources enter the sinusoids where it comes in contact with the hepatocytes. To maximize this contact the sinusoidal surface of the liver is provided with numerous microvilli. Blood from sinusoids drains out through terminal hepatic veins into sublobular and lobular veins and then via medium-sized hepatic veins into large hepatic veins that enters inferior vena cava to reach right atrium. HVOO occur at three major sites in the liver – at the level of the sinusoids, at hepatic veins and at hepatic vein outlets in IVC, which some consider a part of the liver [41]. Of the three primary diseases of HVOO, the site of obstruction to blood flow in SOS is the sinusoids and terminal hepatic vein, in BCS at different level of HVs, and in HVCS at inferior vena cava at HV outlets and at different level of HVs. As hepatic veins have no valves rise in pressure in congestive heart failure and constrictive pericarditis is directly transmitted to hepatic veins resulting in HVOO [42].
Cirrhosis in sinusoidal obstruction syndrome
Sinusoidal obstruction syndrome (SOS), previously known as veno-occlusive disease (VOD) is clinically characterized by hepatomegaly, ascites and jaundice and histologically by ischemic damage of hepatocytes around terminal hepatic veins that shows fibrosis and narrowing. Occurrence of cirrhosis in the disease was recognized with its first report from South Africa. Wilmot and Robertson (1920) who gave an account of the disease from South Africa mentioned that though it was first officially reported in 1918- in a family who developed cirrhosis with ascites after eating bread made from wheat contaminated with seeds of Senecio, it had been prevalent in the country for decades and was recognized locally as ‘bread poisoning’ [43]. The disease was endemic for a long time in central Asian countries Uzbekistan and Tajikistan, where it was described as toxic hepatitis with ascites and known to local inhabitants as ‘camel-belly’ syndrome. Two waves of outbreak of the disease were recorded, the first in 1931-35 with about 1000 to 1500 subjects affected [44] and the second in 1945-46 that affected 60- 70% of the population in agricultural areas [45]. Several accounts of the disease both in children and adults were published in mid 1950s from Jamaica [46]. Brass and colleagues who reported non-portal type cirrhosis among inhabitants of Jamaica who drank herbal tea containing pyrrolizidine alkaloid [47] first named it- veno-occlusive disease. During 1975 to 1977 three outbreaks of the disease was reported from south Asia, two from India and one from Afghanistan [48,49]. Until the advent of chemotherapy in 1950, ingestion of herbal teas or food sources contaminated with PA was the only cause of SOS. Later the disease was reported among patients receiving long term immunosuppressant after renal or liver transplantations [50] and in patients with bone marrow transplantation [51]. De Leve, Shulman and McDonald who observed that the major site of toxic damage in the disease is the hepatic sinusoid renamed it sinusoidal obstruction syndrome in 2004 [39].
VOD was recognized as an important cause of cirrhosis among children in Jamaica. Bras, et al. in 1961 [52] analyzed 1560 autopsy materials from the University College Hospital in Jamaica , of which about a quarter were infant less than one year, mostly from poor black families and detected cirrhosis in 77, of which 30% had evidence of VOD. Development of cirrhosis in PA induced VOD was documented in USA [53]. An important feature of VOD induced cirrhosis is its rapid development usually within 3 months to year of the acute disease. Children were more vulnerable to develop cirrhosis.
Cirrhosis in Budd-Chiari syndrome
Budd-Chiari syndrome is defined as a disease caused by thrombosis of hepatic veins caused by one or several thrombogenic conditions, of which myeloproliferative disorder is the most common. Precipitation of thrombosis of hepatic vein usually requires presence of a combination of thrombogenic conditions and a triggering factor [40]. About 1-3% of the patients with primary myeloproliferative disorders are likely to develop BCS. BCS is a rare disease seen predominantly among Caucasians females of about 35 years with prevalence of 1:100,000 populations [54]. BCS remains asymptomatic unless two or three hepatic veins are obstructed that result in sinusoidal hypertension causing hepatomegaly and ascites. Thrombosis in hepatic veins leads to development of concentric thickening of the vein wall, subintimal fibrosis, development short length stenosis or hepatic vein web. Segment or the whole length of the vein may convert into a fibrous cord like remnant that may ultimately disappear [55].
Tanaka and Wanless [56] who examined 15 resected livers from BCS patients reported common occurrence of thrombosis of portal vein and its branches also. Presence and severity of portal vein thrombosis resulted in different patterns of fibrosis and cirrhosis in BCS. Among the 15 livers six with severe portal vein obliteration from thrombosis had veno-portal cirrhosis, three with normal medium and large portal veins had veno-centric cirrhosis, and the remainder six with moderate portal vein obliteration had veno-centric and venoportal cirrhosis.
Cirrhosis in hepatic vena cava syndrome
The incidence of cirrhosis HVCS in reported series varied from 27 to 100% [12,13,15-19,25,26]. Kage, et al. [57] who studied livers and inferior vena cava from 17 autopsied cases of HVCS observed that hepatic veins of different sizes and portal veins were involved in all, and the occluding lesion or stenosis both in the IVC and intrahepatic veins were due to thrombosis and its squeal. Of the 17 livers, 4 had congestive changes, 7 had extensive congestive fibrosis and the reminder 6 had reversed lobulation or venocentric cirrhosis.
Sinusoidal hypertension
Hepatic microcirculation: Unlike other capillaries hepatic sinusoids have many peculiarities. It is lined by three types of cells, endothelial cells that accounts for 48% of sinusoidal cell population [58], the Kupffer cells and Ito cells. The endothelial cell forms a discontinuous lining of the sinusoid with space between the adjacent cells (Figure 2). It has pores about 100 nm in sizes arranged in sieve plates called fenestrae [59]. There is no basement membrane. The endothelial cells separated by the space of Disse lye almost directly over the pile of carpet of microvilli of the hepatocytes. This arrangement allows free passage of protein and macromolecules but not the formed elements of blood and chylomicrons [58] across the membrane providing opportunity for each liver cell to come in close contact with nutrients and hormones brought by portal vein. To prevent the loss of protein and fluid into the space of Disse the sinusoidal pressure is maintained very low. Whereas the ratio of pre- to post-capillary resistance in skeletal muscle is 4:1, it is 49:1 in the liver sinusoids resulting in a very low outflow resistance [60]. It is further helped by another unique interrelationship between the sinusoidal pressure and the hepatic arterial flow. Increase in sinusoidal pressure results in reflex decrease in hepatic arterial flow [61]. Thus, despite increase in portal venous flow during digestion, the sinusoidal pressure is maintained low. Maintenance of low sinusoidal pressure is important for normal hepatic function.
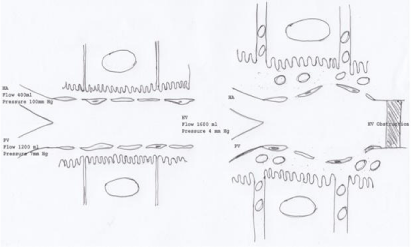
Figure 2: Hepatic sinusoid.
Sinusoidal hypertension: Obstruction to hepatic venous outflow results in sinusoidal hypertension. Increase in sinusoidal pressure brings about a series of changes in the hepatic micro-circulation which results in development of ascites, renal sodium retention, ischemic liver damage and development of liver cirrhosis (Figure 3).
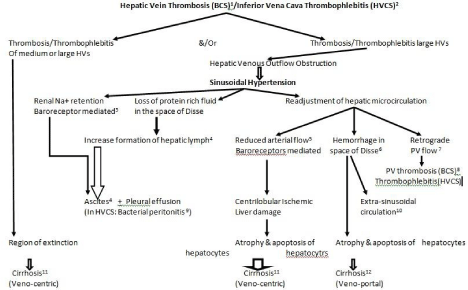
Figure 3: Pathogenesis of LC in sinusoidal hypertension.
Ascites
Sinusoidal hypertension leads to rapid pouring of protein rich fluid into the space of Disse. The fluid is collected by the lymphatics vessels and is carried it back to circulation. Rapid accumulation of the fluid in the space of Disse beyond the capacity of the lymphatic vessel to drain result in oozing out of the fluid on the surface of liver as droplets that coalesces and trickle into the peritoneal cavity. This condition described as ‘weeping liver’ has been frequently observed during peritoneoscopy of the patients with HVCS. This phenomenon was demonstrated by Hyatt, Lawrence and Smith in 1955 in a remarkable series of experiments in dogs by partial constriction of IVC above the opening of HVs [62].
Hepatic baroreceptors mediated renal sodium retention: Increase in sinusoidal pressure causes renal sodium retention mediated by baroreceptors. It often appears before the development of ascites. This was demonstrated by a series of ingenious experiment by Levy and Wexler in dogs where sinusoidal hypertension was induced by partial constriction of supra-hepatic portion of inferior vena cava above [63].
Change in hepatic microcirculation in sinusoidal hypertension: Obstruction of hepatic veins alters the normal circulation of the liver. Blood entering the liver from hepatic artery have to find an alternative pathway to leave liver. Histological study of biopsies and post-mortem tissue conducted by Leopold, Parry and Storring [64] provide evidence extravasation of RBCs occurred into the space of Disse. It was postulated that impairment of nutrition of sinusoidal cells that followed sinusoidal hypertension results to increase in gaps between endothelial cells that allowed egression of RBC into space of Disse where it intermingle with the cells of liver plates resulting in atrophy from compression. It was also postulated that extra sinusoidal circulation made up of capillary size vessels is set up that conducts the blood towards terminal sublobular and large hepatic veins. This extra-luminal circulation continues in a proximal direction to the level necessary to circumvent the obstruction. The other pathway of egression of hepatic artery blood flowing into the sinusoids is through portal vein which develops retrograde flow [65]. In cases where obstruction involves only some of the hepatic veins, the blood drains from obstructed to non-obstructed areas via the portal vein or even via collaterals between hepatic veins [66].
Pathogenesis of Cirrhosis in Sinusoidal hypertension: Sinusoidal hypertension with the extravasation of blood into liver cell plates and the reflex reduction in hepatic arterial inflow combines to cause hemorrhagic necrosis of the hepatocytes around the terminal hepatic venules [1]. The extinct liver cells are replaced by fibrous septa. Liver attempts to maintain its functional capacity by regenerative activity of the hepatocytes around the periportal region. The combined effects ultimately lead to development of veno-centric liver cirrhosis within a few months.
Liver biopsy of patient with acute disease show vascular congestion with loss of hepatocytes near the terminal hepatic vein, atrophic liver cells at mid zone and preserved hepatocytes often with regenerative activity near the portal area. Man and Hall [67] had reported autopsy findings of patients who had died after development of ascites that showed numerous anemic, irregular areas, surrounded by dense red patches. The red areas presented the appearance of passive hyperemia with hemorrhagic extravasation and the pale areas composed of atrophic liver cells, necrotic debris and empty capillaries. Thrombi were observed in many hepatic veins. Microscopically these showed endophlebitic changes and thrombi in various stages of organization with adjacent parenchyma congested and showing changes of early cirrhosis.
In HVOO the portal vein becomes the route of egress with retrograde flow in the vessel. This phenomenon combined with hypercoagulable state in BCS leads to thrombosis in portal vein and its branches; and in HVCS associated with bacterial infection cause thrombophlebitis of portal vein and its branches. Thrombosis or segmental obstruction with thick echoic wall of portal vein wall is commonly observed in ultrasonography and color Doppler examination of patients with HVCS. It had also been observed in autopsy studies of cases of BCS and HVCS [55, 56,57]. Obstruction of portal vein branches leads to development of veno-portal type of liver cirrhosis. Occurrence of veno-portal type of cirrhosis has been documented in BCS [56]. Sinusoidal hypertension is thus the only condition where both veno-centric and veno-portal types of cirrhosis develop.
Features of HVCS induced LC
Cirrhosis in HVCS has many distinctive features [68]. It occur almost equally in both sexes (M:F:: 1.6:1) and it predominantly affects people of younger age group (<50 years). Cirrhosis develops rapidly within several months of occurrence of sinusoidal hypertension. In some patient ascites from HVOO may still be present when liver develop features of cirrhosis (Figure 1). Cirrhosis in HVCS is characterized by relatively preserved hepatocellular function [25,26]. Vascular spiders, palmer erythema and gynaecomastia are uncommon. Clinical picture is dominated by AEs -such as jaundice, mild elevation of ALT/AST or serum bilirubin, or reoccurrence of ascites, associated with neutrophil leukocytosis and high level of CRP. Unlike in alcoholic cirrhosis occurrence of ascites, jaundice and transient esophageal variceal bleeding in HVCS is not due to hepatic decompensation but most often related to severe AE and it responds to treatment with high dose prolonged antibiotics and diuretics. Liver biopsy in such patients shows a mixed picture of cirrhosis and acute congestion [25,26,55]. Patient with cirrhosis due to HVCS who develop jaundice and ascites should not be labeled as de-compensated and pushed to liver transplantation without much workup. Long term prognosis of these patients is good because of the preserved liver function and recurrence of AE may be prevented by improving nutrition and food and water hygiene.
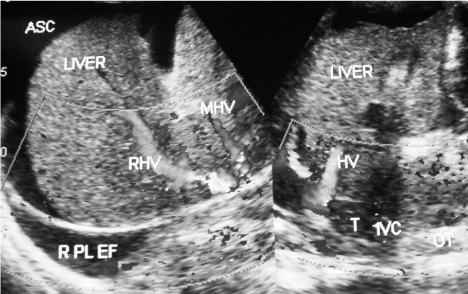
Figure 1: Ultrasonography and Color Doppler of a patient with HVCS who
developed severe acute exacerbation. Hepatic portion of the IVC was filled
with thrombi of different ages causing obstruction to blood flow from all hepatic
veins resulting in hepatomegaly, ascites and right side pleural effusion. Liver
shows the features of liver cirrhosis. (ASC= ascites, RHV= right hepatic vein,
MHV= middle hepatic vein, RPL EF= right side pleural effusion, HV= hepatic
vein, T= recent thrombus, OT= old organized thrombus, IVC= inferior vena
cava).
Beside above mentioned clinical features HVCS related cirrhosis is also characterized by some distinct ultrasonographic and color Doppler (US/CD) features of liver and IVC [68]. Whereas in cirrhosis due to other causes HVs are attenuated, some of the veins may be dilated (Figure 4A). Right inferior hepatic vein may be prominent. Other changes that may be observed include presence of calcified foci in liver related thrombosed HV, segmental stenosis and membrane within and at the outlet of HV, especially of middle hepatic vein. A segment of vein may be thrombosed or replaced by cord like structure or completely lost, with development of collateral (Figure 4B). Commonly right hepatic vein is replaced by a thick cord like structure, or segments of middle and left hepatic veins near the outlet are lost with collateral between the remain patent segments. Changes noted outside the liver include, thick walled gallbladder, thick perihepatitis around the liver and dilated collateral. Hepatic portion of the IVC shows stenosis at cavo-atrial junction with very thick posterior wall and distal segment dilated with thick layer of old organized thrombi of different ages showing capillarization along the posterior wall. IVC at cavo-atrial junction may be completely obliterated. In very chronic patient this site may show bullet-shaped calcification recognized in plane X-Ray film.
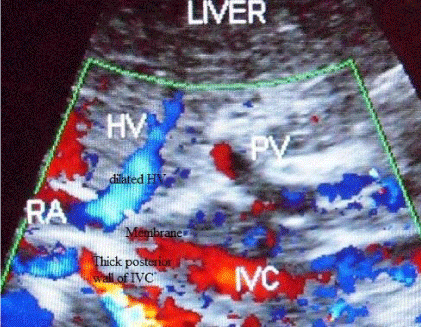
Figure 4A: Ultrasonography and color Doppler of a boy of 8 years with liver
cirrhosis due to HVCS. IVC shows a thick complete obstruction just distal to
the opening of hepatic vein. Posterior wall of the IVC is thick and irregular
projection due to organized thrombus. Hepatic vein is dilated and wall of
portal vein is very thick. (HV= hepatic vein, RA= right atrium, PV= portal vein,
IVC= inferior vena cava).
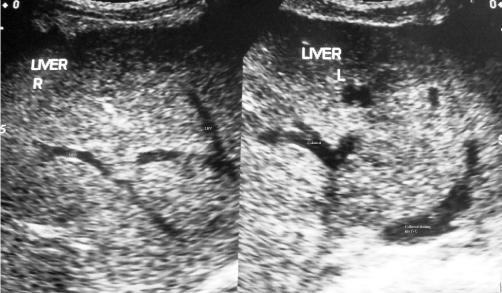
Figure 4B: Ultrasonography of a young lady with cirrhosis due to HVCS.
Long segments of middle and left hepatic veins near the outlet is obliterated
and lost. There is a collateral communication between the remaining patent
segments that drain into IVC through a collateral.
Long term follow-up of patients with cirrhosis due to HVCS showed that it was complicated by moderate incidence of HCC of 10% [12]. Development of HCC was related to long duration of the disease and increased frequency of acute exacerbations. As acute exacerbation is precipitated by bacterial infection it may be possible to prevent HCC in patients with HVCS by adopting a policy of zero tolerance to bacterial infection.
HVCS was frequently reported from Japan in early 20th century [15-19] and from Europe and North America in 19th and 20th century [69-74]. This disease has now almost disappeared from these areas. It is now reported only occasionally rarely from these areas [75-79]. As HVCS is caused by bacterial infection and it associated with high incidence of cirrhosis it is possible that the disease may be common in developing countries but has not received due attention. In the past HVCS has remained an underdiagnosed entity in the area [80]. Incidence of cirrhosis of unknown etiology is very high in Asian countries [9,81-82]. Some of it may be related to HVCS. As treatment and prognosis of HVCS related cirrhosis is different [68] it is suggested that an attempt should be made to identify the exact cause of cirrhosis in these patients. Routine use of ultrasonography and color Doppler examination of the inferior vena cava and liver in persons with bacterial infection and patient with cirrhosis is suggested to assess its prevalence in the developing countries.
Conclusion
Development of sinusoidal hypertension is a dramatic event in the natural history of the disease related to HVOO. In HVCS it is precipitated by bacterial infection. Sinusoidal hypertension leads to development of ascites, portal hypertension and severe ischemic liver damage, and is rapidly followed by development of cirrhosis within a few months. Cirrhosis due to sinusoidal hypertension is characterized by distinctive features. It occurs in any age group including children with equally prevalence in both sexes. It is characterized by presence of distinctive US/CD features of liver and inferior vena cava and rarity of signs like palmer erythema and vascular spiders. Jaundice, ascites and transient bleeding from esophageal varices occur due to acute exacerbation precipitated by bacterial infection that responds to medical treatment. These signs do not indicate hepatic decompensation. Cirrhosis due to sinusoidal hypertension has better prognosis due to preserved hepatocellular function. Hepatic vena cava syndrome may be a common cause of cirrhosis in developing countries where it occur alone or as co-morbid condition in patients with alcohol abuse or chronic hepatitis B or C.
References
- Wanless IR. Pathogenesis of cirrhosis. J Gastroenterol & Hepatol. 2004; 19: S369-S371.
- Herrick FC. An experimental study into the cause of increased portal pressure in portal cirrhosis. J Exp Med. 1907; 9: 93-104.
- McIndoe AH. Vascular lesions of portal cirrhosis. Ach Pathol. 1928; 5: 23-42.
- Kelty RH, Baggenstoss AH, Butt HR. The relation of the regenerated hepatic nodule to vascular bed in cirrhosis. Proc Staff Meetings Mayo Clin. 1950; 25: 17-26.
- Michitaka K, Nishiguchi S, Aoyagi Y, Hiasa Y, Tokumoto Y, Onji M et al. Etiology of liver cirrhosis in Japan: a nationwide survey. J Gastroenterol. 2009; 45: 86-94.
- Yoshizawa H. Hepatocellular carcinoma associated with hepatitis C virus infection in Japan: projection to other countries in the foreseeable future. Onclology. 2002: 62: 8-17.
- Bras G, Brooks SE, Watler DC. Cirrhosis of liver in Jamaica. J Pathol Bacteriol. 1961; 82: 503-512.
- Nayak NC. Indian childhood cirrhosis. In: Ahuja MMS, editor. Progress in clinical medicine, Second series. Arnold-Heinnemann. 1978; 304-314.
- Shrestha SM, Tsuda F, Okamoto H, T Tanaka, Y Miyakawa, M Mayumi, et al. Hepatitis B virus subtypes and hepatitis C virus genotypes in patients with chronic liver disease in Nepal. Hepatology. 1994; 19: 805-809.
- Shrestha SM. Shrestha S. Shrestha A, Tsuda F, Endo K, Takahashi M, et al. High prevalence of hepatitis B virus infection and inferior vena cava obstruction among patients with liver cirrhosis or hepatocellular carcinoma in Nepal. J Gastroenterol & Hepatol. 2007; 22: 1921-1928.
- Shrestha SM, Okuda K, Uchida T, Maharjan KG, Shrestha S, Joshi BL, et al. Endemicity and clinical picture of liver disease due to obstruction of the hepatic portion of the inferior vena cava in Nepal. J Gastroenterol Hepatol. 1996; 11: 170-179.
- Shrestha SM. Liver cirrhosis and hepatocellular carcinoma in hepatic vena cava disease, a liver disease caused by obstruction of inferior vena cava. Hepatol Int. 2009; 3: 392-402.
- Shrestha SM, Shrestha S. Hepatic vena cava syndrome: a common cause of liver cirrhosis in children in Nepal. Trop Gastroenterology. 2014; 35: 85-95.
- Janssen HL, Garcia-Pagan JC, Elias E, Mentha G, Hadengue A, Valla DC. European Group for the Study of Vascular Disorders of the Liver. Budd-Chiari syndrome: a review by an expert panel. J Hepatol. 2003; 38: 364-371.
- Nakamura T, Nakamura S, Aikawa T, Suzuki O, Onodera A, Karoji N, et al. Obstruction of the inferior vena cave in the hepatic portion and the hepatic veins: report of eight cases and review of the Japanese literature. Angiology. 1968; 19: 479-498.
- Yamamoto S, Yokoyama Y, Takeshige K, Iwatsuki S. Budd-Chiari syndrome with obstruction of the inferior vena cava. Gastroenterology. 1968; 54: 1070- 1084.
- Takeuchi J, Takada A, Hasumura Y, Matsuda Y, Ikegami P. Budd-Chiari syndrome associated with obstruction of the inferior vena cava. A report of seven cases. Am J Med. 1971; 51: 11-20.
- Nakamura S, Takezawa Y. Obstruction of the inferior vena cava in the hepatic portion and hepatocellular carcinoma. Tohoku J Exp Med. 1982; 138: 119- 120.
- Ono J, Sakoda K, Kawada T. Membranous obstruction of the inferior vena cava. Ann Surg. 1983; 197: 454-458.
- Lee BB, Villavicencio L, Kim Y W, Do YS, Koh KC, Lim HK, et al. Primary Budd-Chiari syndrome: outcome of endovascular management for suprahepatic venous obstruction. J Vas Surg. 2006; 43: 101-108.
- Wang ZG. Management of Budd-Chiari syndrome: experience from 430 cases. Asian J Surg. 1996; 19: 23-30.
- Wang ZG, Zhang FJ, Li XQ, Meng QY. Management of Budd-Chiari syndrome: what is the best approach? J Gastroenterol & Hepatol. 2004; 19: S212-S218.
- Datta DV, Saha S, Singh SA, Gupta BB, Aikat BK, Chutani PN. Clinical spectrum of Budd-Chiari syndrome in Chandigrah with particular reference to obstruction of intrahepatic portion of inferior vena cava. Indian J Med Res. 1972; 60:385-401.
- Madangopalan N, Solomon V, Jayanthi V, Raghuram K, Balakumar M, Kandasway I, et al. Clinical spectrum of chronic Budd-Chiari syndrome and surgical relief for ‘coarctation’ of the inferior vena cava. J Gastroenterol and Hepatol. 1986; 1: 359-369.
- Dilawari JB, Bambery P, Chawla Y, Kaur U, Bhusnurmath SR, Malhotra HS, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of literature. Medicine. 1994; 73: 21-36.
- Singh V, Sinha SK, Nain CK, Bambery P, Kaur U, Verma S, et al. Budd-Chiari syndrome: our experience of 71 patients. J Gastroenterol Hepatol. 2000; 15: 550-554.
- Simson IW. Membranous obstruction of the inferior vena cava and hepatocellular carcinoma in South Africa. Gastroenterology. 1982; 82: 171- 178.
- Kew MC, McKnight A, Hodkinson J, Bukofzer S, Esser JD. The role of membranous obstruction of the inferior vena cava in the etiology of hepatocellular carcinoma in Southern African Blacks. Hepatology. 1989; 9: 121-125.
- Awwad S. The Budd-Chiari syndrome. J Egypt Med Assoc. 1952; 35: 650- 669.
- Okuda K. Inferior vena cava thrombosis at its hepatic portion (obliterative hepatocavopathy). Semin Liver Dis. 2002; 22: 15-26.
- Shrestha SM, Shrestha S. Hepatic vena cava disease: etiologic relation to bacterial infection. Hepatology Research. 2007; 37: 196-204.
- Shrestha SM, Kage M, Lee BB. Hepatic vena cava syndrome: New concept of pathogenesis. Hepatol Res. 2017; 47: 603-615.
- Pleasants JH. Obstruction of inferior vena cava with a report of 18 cases. Johns Hopkins Hospital Rpt 1911; 16: 363-546.
- Shrestha SM. Pleural effusion in hepatic vena cava disease. Kathmandu Univ Med J (KUMJ). 2007; 5: 218-224.
- Shrestha SM, Shrestha S. Bacterial peritonitis in the hepatic inferior vena cava disease: a hypothesis to explain the cause of infection in high protein ascites. Hepatology Res. 2002; 24: 42-49.
- Khuroo MS, Datta DV. Budd-Chiari syndrome following pregnancy. Report of 16 cases, with roentgenologic, hemodynamic and histologic studies of the hepatic outflow tract. Am J Med. 1980; 68: 113-121.
- Shrestha SM, Ghimire RK, Basnyat P, Pradhan V, Poudel V. Acute on chronic phenomenon in hepatic IVC obstruction: a case report. Trop Gastroenterol. 1999; 20: 182-184.
- Shrestha SM. Diagnosis of Hepatic Vena Cava Syndrome by ultrasonography and color Doppler based on new concept of its pathogenesis. EC Gastroenterology and Digestive System 2017; 2: 256-270.
- De Leve LD, Shulman HM, McDonald GB. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (Veno-occlusive disease). Seminar in Liver Disease. 2002; 22: 27-41.
- Valla DC. Hepatic vein thrombosis (Budd-Chiari syndrome). Semin Liver Dis. 2002; 22: 5-14.
- Ludwig J, Hashimoto E, McGill DB, Heerden JA. Classification of hepatic venous outflow obstruction: ambiguous terminology of the Budd-Chiari syndrome. Mayo Clin Proc. 1990; 65: 51-55.
- Arora A, Tandon N, Sharma MP, Acharya SK. Constrictive pericarditis masquerading as Budd-Chiari syndrome. J Clin Gastroenterol. 1991; 13: 178-181.
- Wilmot FC, Robertson GW. Senecio disease or cirrhosis of liver due to Senecio poisoning. Lancet. 1920; 848-849.
- Mirochnik MF, ed: Functional, diagnostic and pathological changes of toxic hepatitis with ascites. Tashkent, State Publishing House of Science Technology and Socio-economic Literature of Uzbekistan (in Russian).
- Ismailov NI. Heliotropic toxicosis (toxic hepatitis with ascites). Academy of Sciences of Uzbekistan, Tashkent. 1948: 1-120.
- Jelliffe DB, Brass G, Stuart KL. Veno-occlusive disease of the liver Pediatrics 1954;14: 334-339
- Bras G, Jelliffe DB, Stuart KL. Veno-occlusive disease of liver with non portal type of cirrhosis, occurring in Jamaica. AMA Arch Pathol. 1954; 57: 285-300.
- Mohabbat O, Younos MS, Merzad AA, Srivastava RN, Sediq GG, Aram GN. An outbreak of hepatic veno-occlusive disease in north-western Afghanistan. Lancet. 1976; 2: 269-271.
- Tandon RK, Tandon BN, Tandon HD, Bhatia ML, Bhargava S, Lal P, et al. Study of an epidemic of venoocclusive disease in India. Gut. 1976; 17: 849- 855.
- Read AE, Wiesner RH, LaBrecque DR, Tifft JG, Mullen KD, Sheer RL, et al. Hepatic veno-occlusive disease associated with renal transplantation and azathioprine therapy. Ann Intern Med. 1986; 104: 651-655.
- Berk PD, Popper H, Krueger GR, Decter J, Herzig G, Graw RG Jr. Venoocclusive disease of the liver after allogeneic bone marrow transplantation: possible association with graft-versus-host disease. Ann Intern Med. 1979; 90: 158-164.
- Bras G, Brooks SE, Watler DC. Cirrhosis of the liver in Jamaica. J Pathol Bacteriol. 1961; 82: 503-512.
- Stillman AS, Huxtable R, Consroe P, Kohnen P, Smith S. Hepatic venoocclusive disease due to pyrrolizidine (Senecio) poisoning in Arizona. Gastroenterology. 1977; 73: 349-352.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis. 2008; 28: 259- 269.
- Parker RG. Occlusion of the hepatic veins in man. Medicine (Baltimore). 1959; 38: 369-402.
- Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndrome: portal vein thrombosis and histogenesis of veno-centric cirrhosis, veno-portal cirrhosis regenerative nodules. Hepatology. 1998; 27: 488-496.
- Kage M, Arakawa M, Kojiro M, Okuda K. Histopathology of membranous obstruction of the inferior vena cava in the Budd-Chiari syndrome. Gastroenterology. 1992; 102: 2081-2090.
- Bradfield JW. Liver sinusoidal cells. J Pathol. 1984; 142: 5-6.
- Lautt WW. Hepatic vasculature: a conceptual review. Gastroenterology. 1977; 73: 1163-1169.
- Greenway CV, Stark RD. Hepatic vascular bed. Physiol Rev. 1971; 51: 23-65.
- Hyatt RE, Lawrence GH, Smith JR. Observations on the origin of ascites from experimental hepatic congestion. J Lab Clin Med. 1955; 45: 274-280.
- Levy M, Wexler MJ. Sodium excretion in dogs with low-grade caval constriction: role of hepatic nerves. Am J Physiol. 1987; 253: F672-F678.
- Leopold JG, Parry TE, Storring FK. A change in the sinusoid-trabecular structure of the liver with hepatic venous outflow block. J Path. 1970; 100: 87-98.
- Pollard J, Nebesar R. Altered hemodynamic in the Budd-Chiari syndrome demonstrated by selective hepatic and selective spleenic angiography. Radiology. 1967; 89: 236.
- Maguire R, Doppman JL. Angiographic abnormalities in partial Budd-Chiari syndrome. Radiology. 1977; 122: 629-635.
- Mann JD and Hall IW. Obstruction of the inferior vena cava. Edinburgh MJ. 1904; 16: 56-62.
- Shrestha SM. Liver cirrhosis in hepatic vena cava syndrome (or membranous obstruction of inferior vena cava). World J Hepatol. 2015; 7: 874-884.
- Wilks S. Obstruction of hepatic veins and vena cava by fibrous deposit in the liver. Trans Path Soc London. 1862; 13:122-124.
- Osler W. Obliteration of Vena Cava Inferior, with great Stenosis of Orifices of Hepatic Veins. J Anat Physiol. 1879; 13: 291-304.
- Willcocks F. Perihepatitis; thrombosis of inferior vena cava at point of entry of hepatic veins; ascites and great varicosity of superficial veins over anterior thoracic and abdominal parietes. Trans Path Soc London. 1896; 47: 67-68.
- Thompson T, Turnbull HM. Primary occlusion of the ostia of the hepatic veins. Quart. J Med 1912; 5: 277-295.
- Hutchison R, Simpson SL. Occlusion of the Hepatic Veins with Cirrhosis of the Liver. Arch Dis Child. 1930; 5: 167-186.
- Rigdon RH. On the relation between the thrombophlebitis of the inferior vena cava and occlusion of the hepatic veins. Bull Johns Hopkins Hosp. 1933; 53:162-171.
- Okuda H, Yamagata H, Obata H, Iwata H, Sasaki R, Imai F, et al. Epidemiological and clinical features of Budd-Chiari syndrome in Japan. J Hepatol. 1995; 22: 1-9.
- Angelman H, Spencer R. The syndrome of obstruction of inferior vena cava in childhood. Br Med J. 1950; 2: 752-755.
- Kibel MA, Marsden HB. Inferior vena caval and hepatic vein thrombosis: the Chiari syndrome in childhood. Arch Dis Child. 1956; 31: 225-228.
- Mc Clead RE, Birken G, Wheller JJ, Hansen, Nancy B, Bickers, et al. Budd- Chiari syndrome in a premature infant receiving total parenteral nutrition. Pediatric Gastroenterology and Nutrition. 1986; 5: 655-658.
- Lois JF, Hartzman S, McGlade CT, Gomes AS, Grant EC, Berquist W, et al. Budd-Chiari syndrome: Treatment with percutaneous transhepatic recanalization and dilation. Radiology. 1989; 170: 791-793.
- Shrestha SM. Membranous obstruction of the hepatic portion of the inferior vena cava: is this an underdiagnosed entity in developing countries? Am J Gastroenterol. 1995; 90: 303-306.
- Hadiwandowo S, Tsuda F, Okamoto H, Tokita H, Wang Y, Tanaka T, et al. Hepatitis B virus subtypes and hepatitis C virus genotypes in patients with chronic liver disease or on maintenance hemodialysis in Indonesia. J Med Virol. 1994; 43: 182-186.
- Luengrojanakul P, Vareesangthip K, Chainuvati T, Murata K, Tsuda F, Tokita H, et al. Hepatitis C virus infection in patients with chronic liver disease or chronic renal failure and blood donors in Thailand. J Med Virol. 1994; 44: 287-292.