
Case Report
Ann Hematol Oncol. 2016; 3(10): 1118.
A Case of Paroxysmal Cold Hemoglobinuria with Subsequent Progression to Acute Myelogenous Leukemia
Ibrahim M*, Ogunleye F, Stender M, Jaiyesimi I and Huben M
Department of Hematology and Oncology, Oakland University William Beaumont School of Medicine, USA
*Corresponding author: Mohammed Ibrahim, Department of Hematology and Oncology, Oakland University William Beaumont School of Medicine, Royal Oak, Michigan 48073, USA
Received: September 19, 2016; Accepted: October 18, 2016; Published: October 21, 2016
Abstract
Paroxysmal cold hemoglobinuria is a rare hematologic condition that causes autoimmune complement-mediated hemolysis on exposure to cold temperatures. The spectrum of presentation can range from a self-limited postviral hemolysis to a life-threatening acute hemolytic anemia. Timely recognition of this condition is critical, given the associated morbidity and mortality. We report a case of a 65 year-old male who presented to the hematology clinic with anemia and fatigue. Laboratory investigations revealed macrocytic anemia with elevated lactate dehydrogenase level, low haptoglobin and the presence of urine hemosiderin, raising concern for intravascular hemolysis. Testing for Donath Landsteiner antibody was positive, confirming the diagnosis of paroxysmal cold hemoglobinuria. Supportive care measures were instituted and the patient was told to avoid cold exposure. The patient eventually developed acute myelogenous leukemia and succumbed to the disease. It is important for physicians to be vigilant to the possibility of coexisting or subsequent malignancies in patients diagnosed with paroxysmal cold hemoglobinuria.
Keywords: Paroxysmal cold hemoglobinuria; PCH; Hemolysis; Anemia; Acute myelogenous leukemia; AML
Introduction
Paroxysmal Cold Hemoglobinuria (PCH) is a rare cause of autoimmune complement-mediated hemolytic anemia. In 1872, a syndrome with the occurrence of red colored urine after exposure to cold temperature was described. The antibody causing PCH was first reported by Donath and Landsteiner in 1904 [1]. PCH causes a hemolytic anemia due to a cold-reacting polyclonal IgG antibody that binds to “P” antigen on the surface of the Red Blood Cell (RBC) in cold temperature and fixes complement. When the RBCs are subsequently warmed to 37°C, the complement cascade is activated, causing intravascular hemolysis, hemoglobinuria and hemosiderinuria [2].
Most cases of PCH are seen in children, where it accounts for 1-5% of childhood Autoimmune Hemolytic Anemia (AIHA) cases. In one review of 52 cases of PCH, the age range was 1 to 82 years with a median age of 5 years of age. Sokol, et al. [3] have estimated that the annual incidence of PCH is 0.4 cases per 100,000 people. The most common origins of PCH are post-infectious (eg., measles, mumps, Epstein-Barr virus (EBV), influenza, Mycoplasma pneumoniae) and autoimmune (related to lymphomas, Chronic lymphocytic leukemia (CLL), myelofibrosis, myelodysplastic syndrome and small cell lung cancer) [4-8]. Association with tertiary syphilis has been well described, as a syphilis antigen generates a cross-reacting antibody that causes this syndrome.
The adult form of PCH is usually chronic, lasting several years. Clinical features include presence of dark colored urine (hemoglobinuria) beginning several minutes to hours after exposure to cold, as well as Raynaud’s phenomenon, back pain, leg pains, abdominal pain, chills and fever, and general malaise related to the release of large quantities of hemoglobin from lysed RBCs, which then act as an irritant to various tissues. Laboratory investigations demonstrate evidence of intravascular hemolysis with jaundice, reticulocytosis, hemosiderinuria, elevated indirect bilirubin, elevated lactate dehydrogenase (LDH) level, low haptoglobin and low complement. The hemolysis can be acute and massive. Spherocytes and erthrophagocytes may be seen on the peripheral smear [9,10]. Direct Antiglobulin Test (DAT) can be positive for anti C3 and negative for IgG during the hemolytic episode.
Diagnosis is made using the Donath Landsteiner test. The patient’s serum is incubated with normal RBCs and pooled human serum (as a source for complement) at 40C, which allows the antibody and the early components of complement to be fixed, and then at 37°C for later components of complement to be activated [1,11]. Hemolysis occurs if the antibody is present.
Treatment of PCH includes supportive care to keep patient warm and avoiding exposure to cold. If life-threatening anemia occurs, transfusion of packed RBCs through a blood-warming device is needed. Most patients develop iron deficiency and require iron replacement. In general, the prognosis of PCH is good with most patients responding to supportive measures and treatment of any underlying condition [3]. There has been one case report of an 18-yearold female who was diagnosed with PCH during pregnancy. With supportive measures alone, hemolysis resolved within 6 weeks and she had a successful pregnancy outcome with no evidence of hemolysis or anemia in the newborn [12]. Anecdotal reports have indicated the use of steroids (oral prednisone at a dose of 1 mg/kg) to be beneficial if symptoms are severe and/or life-threatening. For chronic episodes, immunosuppression with prednisone, cyclophosphamide and/or azathioprine have been tried [11]. Koppel, et al. [13] described a case of a 64 year old female with steroid-refractory PCH who was treated with rituximab 375 mg/m2 weekly for 4 doses and had a normalization of her hemoglobin and cessation of hemolysis. When the hemolytic anemia recurred after 9 months, this patient was re-challenged with the same regimen of rituximab and again had a cessation of hemolysis and normalization of her hemoglobin. She remained in remission at 19 month follow-up. Eculizumab has been tried in 1 patient with PCH associated with multiple myeloma, but the patient did not have a response [14]. Given the rarity of this condition, it is unlikely that prospective clinical trials will ever be conducted. There is no role for splenectomy as hemolysis is intravascular.
The main differential diagnosis of PCH is cold agglutinin disease. This is associated with agglutination of red blood cells on the peripheral smear, elevated titers of Ig M cold agglutinin, but the Donath Landsteiner test is negative (Table 1).
![]()
PCH
Cold agglutinin disease
Donath Landsteiner test
+++
-
Cold agglutinin titers Ig M
+
+++
RBC agglutination
-
+++
Table 1: Differences between PCH and cold agglutinin disease.
Case Presentation
A 64 year-old male was referred to a hematologist by his Primary Care Physician for evaluation of fatigue and anemia. He had no history of GI bleeding or blood in the urine. He denied chest pain, shortness of breath or palpitations. On examination, his blood pressure was 142/80 mm Hg, heart rate was 82/min, respiratory rate was 12/min and temperature was 96.8F. There were no jaundice, lymphadenopathy or splenomegaly on examination. Laboratory investigations showed a hemoglobin of 11.3 g/dl (normal range: 13.5- 17.0 g/dL) with an MCV of 110 fL (normal range 80-100 fL), platelet count of 135 bil/L (normal range 150-400 bil/L) and a White Blood Cell (WBC) count of 5.7 bil/L (normal range: 3.5-10.1 bil/L). The peripheral smear showed a moderate hypochromic anemia with the presence of spherocytes (Figure 1). Given the absence of agglutination on the peripheral smear, cold agglutinin disease was felt to be unlikely and therefore cold agglutinin testing was not performed. Iron levels, vitamin B-12 and folic acid levels were found to be within normal limits. The LDH level was elevated at 253 U/L (normal range: 100- 238 U/L) and haptoglobin was less than 8 mg/dl (normal range: 40- 240 mg/dL). The Direct Antiglobulin Test (DAT) was negative. His urine tested positive for hemosiderin. Liver enzymes were normal including a normal total bilirubin of 0.4 (normal range: 0.3-1.2 mg/dl). Due to the presence of elevated LDH, low haptoglobin and urine hemosiderin, there was concern for intravascular hemolysis. Flow cytometry for paroxysmal nocturnal hemoglobinuria (PNH) was negative. A Donath Landsteiner antibody test was positive, confirming the diagnosis of paroxysmal cold hemoglobinuria. The patient had a Venereal Disease Research Laboratory (VDRL) test which was negative.
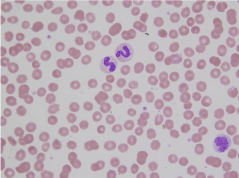
Figure 1: Peripheral smear showing hypochromic RBCs and presence of
spherocytes (Wright Giemsa stain 100 x magnification).
Supportive measures were initiated. The patient was placed on oral iron as well as folic acid therapy and was advised to avoid exposure to cold conditions and to wear warm clothing, especially during the winter. He did not require hospitalization. The patient had improvement in his hemoglobin with supportive measures alone, but then developed a mild chronic anemia over the next two years (Figure 2). LDH levels also improved with supportive measures (Figure 3).
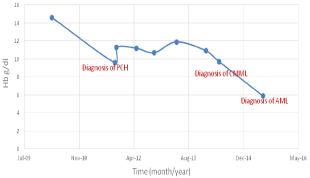
Figure 2: Trend in hemoglobin (Hb) levels prior to and after diagnosis of
PCH.
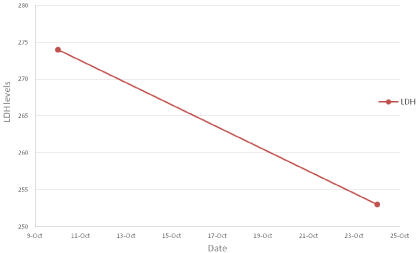
Figure 3: Improvements in LDH levels with supportive care and avoidance
of cold.
Two and a half years after being diagnosed with PCH, the patient was noted to have worsening anemia with development of a monocytosis. A bone marrow biopsy revealed Chronic Myelomonocytic Leukemia (CMML). He was placed on active surveillance. One year after being diagnosed with CMML, he had an acute drop in hemoglobin to 7 g/dl (Figure 2) with blasts in the peripheral smear. A repeat bone marrow biopsy revealed Acute Myelogenous Leukemia (AML). Blood transfusions were given using a blood warming device to avoid hemolysis. The patient was treated with standard induction therapy with 7 days of Cytarabine and 3 days of Idarubicin. He received consolidation therapy with high-dose Cytarabine, and underwent a matched, unrelated allogenic stem cell transplant. Unfortunately he developed recurrence of his leukemia and died from the disease.
Discussion
This case highlights the unique characteristics of PCH presentation in adults. A high index of suspicion should be maintained for PCH in any patient presenting with evidence of intravascular hemolysis. Our patient had a rapid improvement in his hemoglobin with supportive care measures alone. However, he subsequently had a mild chronic anemia that was likely related to slow ongoing hemolysis. It is unclear if he complied completely with instructions regarding avoiding exposure to cold. Most patients with PCH have evidence of iron deficiency. Our patient had normal iron levels at diagnosis because he was taking iron supplements. Most patients with PCH also have a positive DAT at least for C3/C3d, however our patient had a negative DAT. DAT-negative autoimmune hemolytic anemias (AIHAs) account for 5% to 10% of all AIHAs. In a review by Garratty [15], three causes of a negative DAT were identified including (1) small numbers of red blood cell (RBC)-bound IgG molecules that are below the threshold of the DAT; (2) IgA and IgM autoantibodies and (3) lowaffinity autoantibodies. We suspect the DAT was negative for C3 in our patient because complement was depleted as a result of significant intravascular hemolysis. Our patient was later diagnosed with CMML and then with AML. The association of PCH with hematological malignancies such as lymphomas, chronic lymphocytic leukemia, or myelodysplastic syndrome has been well described [4-7]. A case of PCH associated with small cell lung cancer has also been published [8]. Physicians need to be vigilant about the possibility of coexisting or subsequent malignancies in patients diagnosed with PCH.
References
- Donath J, Landsteiner K. Über Paroxysmale Hämoglobinurie. Münchener medizinische Wochenschrift. 1904; 51: 1590-1593.
- Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Ann N Y Acad Sci. 1965; 124: 456-461.
- Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: Paroxysmal cold haemoglobinuria: A clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999; 4: 137-164.
- Sharara AI, Hillsley RE, Wax TD, Rosse WF. Paroxysmal cold hemoglobinuria associated with non-Hodgkin's lymphoma. South Med J. 1994; 87: 397-399.
- Sivakumaran M, Murphy PT, Booker DJ, Wood JK, Stamps R, Sokol RJ. Paroxysmal cold haemoglobinuria caused by non- hodgkin's lymphoma. Br J Haematol. 1999; 105: 278.
- Breccia M, D'Elia, GM, Girelli G, Vaglio S, Gentilini F, Chiara S, Alimena G. Paroxysmal cold haemoglobinuria as a tardive complication of idiopathic myelofibrosis. European Journal of Haematology. 2004; 73: 304-306.
- Stefanizzi C, Breccia M, Santopietro M, Coluzzi S, Cannella L, Girelli G, et al. Unusual association of paroxysmal cold hemoglobinuria as the first sign of disease in myelodysplastic patient. Int J Hematol. 2009; 89: 720-721.
- Lippman SM, Winn L, Grumet FC, Levitt LJ. Evans' syndrome as a presenting manifestation of atypical paroxysmal cold hemoglobinuria. Am J Med. 1987; 82: 1065-1072.
- Hernandez JA, Steane SM. Erythrophagocytosis by segmented neutrophils in paroxysmal cold hemoglobinuria. Am J Clin Pathol. 1984; 81: 787-789.
- Mukhopadhyay S, Keating L, Souid AK. Erythrophagocytosis in paroxysmal cold hemoglobinuria. Am J Hematol. 2003; 74: 196-197.
- M. A. Gertz. Cold hemolytic syndrome. Hematology/The Education Program of the American Society of Hematology. 2006; 19-23.
- Akpoguma AO, Carlisle TL, Lentz SR. Case report: paroxysmal cold hemoglobinuria presenting during pregnancy. BMC Hematology. 2015; 15: 3.
- Koppel A, Lim S, Osby M, Garratty G, Goldfinger D. Rituximab as successful therapy in a patient with refractory paroxysmal cold hemoglobinuria. Transfusion. 2007; 47:1902.
- Gregory GP, Opat S, Quach H, Shortt J, Tran H. Failure of eculizumab to correct paroxysmal cold hemoglobinuria. Ann Hematol. 2011; 90: 989.
- Garratty G. Immune Hemolytic Anemia Associated with Negative Routine Serology. Seminars in Hematology. 42: 156 -164.