
Case Report
Ann Hematol Oncol. 2017; 4(2): 1136.
Using Next-Generation Sequencing to Find Targetable Genomic Lesions in a Case of Relapsed Extramedullary Acute Myeloid Leukemia
Miller KC, Dias AL, Patnaik MM and Litzow MR*
Division of Hematology, Mayo Clinic, USA
*Corresponding author: Litzow MR, Division of Hematology, Mayo Clinic, Rochester, MN, 200 First Street SW, Rochester, MN 55905, USA
Received: January 15, 2017; Accepted: February 17, 2017; Published: February 20, 2017
Abstract
Next-generation sequencing (NGS) has catalyzed a revolution in our understanding and treatment of leukemia, in some cases revealing crypticgenomic alterations that can be specifically targeted with drugs, such as tyrosine kinase inhibitors (TKIs). Herein we describe a case of relapsed extramedullary acute myeloid leukemia (AML), for which NGS of a tumor biopsy revealed several genomic lesions, including an uncommon ETV6-ABL1 fusion oncogene. Consequently, we began treatment with the TKI dasatinib, in combination with 5-azacitidine. This report demonstrates the clinical value of using NGS to find targeted therapies for patients with hematological malignancies such as AML.
Keywords: Extramedullary acute myeloid leukemia; Myeloid sarcoma; ETV6-ABL1; Dasatinib; Next-generation sequencing (NGS)
Introduction
Next-generation sequencing (NGS) technology is rapidly evolving, and has begun to unravel the unique complexities of hematologic malignancies. Using this technology, there has been a recent, robust effort to parse hematologic diseases such as acute myeloid leukemia (AML) into subgroups that have both prognostic and therapeutic value, with the goal of improving patient outcomes [1].
Moreover, advances in both technological capacity and bioinformatics have made it feasible to use NGS to inform therapy for individual patients, often revealing unique genetic lesions that can be specifically targeted with drugs, such as tyrosine kinase inhibitors (TKIs). In this case, we describe a patient with relapsed myeloid sarcoma to illustrate how NGS was used to identify a rare fusion gene that was ultimately targeted with a TKI.
Case Presentation
A previously healthy 42-year-old white woman presented with right-sided back pain in February 2015. She was treated symptomatically. Four months later, she returned with persistent pain, exertional dyspnea and facial swelling. A CT scan revealed a large anterior mediastinal mass (11x6 cm) and significant bilateral pleural effusions.
An initial CBC was relatively normal: Hgb 10.9 g/dL; RBC 4.01x1012/L; MCV 85.3 fL; WBC 11.83x109/L; PLT 437x109/L. The peripheral smear had 92% neutrophils, 6% lymphocytes, and 2% monocytes, with no morphologic abnormalities. Additionally, a comprehensive metabolic panel was normal except for an elevated lactate dehydrogenase, which was 307 U/L (upper limit of normal is 245 U/L). A CT-guided biopsy of the mediastinal mass demonstrated a myeloid sarcoma with monocytic differentiation, which was positive for CD68, CD34 (dim, partial), CD43, CD45 (dim), CD4 (dim, variable), and myeloperoxidase (partial). Ki67 proliferative staining was high at approximately 90%. Bone marrow (BM) biopsy demonstrated 4% myeloid blasts. Cytogenetic studies from the BM aspirate showed complex cytogenetic abnormalities with six out of 20 metaphases demonstrating +8, +20, deletion 9p, deletion 16p, and a balanced translocation between chromosomes 1 and 11. Fluorescence in situ hybridization (FISH) confirmed a deletion of the CREBBP gene region at 16p13.3, consistent with the 16p deletion. Staging positron emission tomography (PET) scan revealed an FDG-avid mediastinal mass, with a standard uptake value (SUV) maximum of 11.8 (Figure 1), paratracheal/superior-mediastinal lymph nodes and a single anterior iliac wing bone lesion. Peripheral blood testing for FLT3 mutations was negative. Spinal fluid analysis was negative for leukemic involvement.
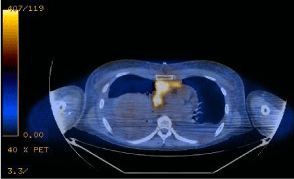
Figure 1: PET-CT at the time of disease presentation demonstrated a
mediastinal mass, which was 11 x 6 cm with a standard uptake value (SUV)
maximum of 11.8, paratracheal/superior-mediastinal lymph nodes and a
single anterior iliac wing bone lesion.
She was diagnosed with myeloid sarcoma with minimal BM involvement, and started on induction chemotherapy for highrisk acute myeloid leukemia with idarubicin (IDA; 12 mg/m2/day for 3 days) and high-dose cytarabine (HiDAC; 1500 mg/m2/day by continuous IV infusion for 4 days). Post-induction complications included culture-negative neutropenic fevers, severe anorexia, mild gastrointestinal symptoms, drug-induced rash (likely due to cytarabine),as well as bilateral subclavian and axillary deep vein thromboses.
A follow up PET scan showed marked regression of her myeloid sarcoma and she was referred for hematopoietic stem cell transplantation (HSCT). Her sister was found to be a 10/10 human leukocyte antigen (HLA) match. Meanwhile, she underwent two rounds of high-dose cytarabine consolidation (3 g/m2 b.i.d. on days 1, 3 and 5). Her pre-transplant PET scan showed new metabolically active adenopathy involving the anterior mediastinum, left internal mammary lymph nodes, right hilum, and a new 1.8cm nodule near the ascending aorta (SUV max 11.2). Subsequently, transplant was delayed and she was treated with salvage CLAG-M [cladribine 5 mg/ m2/day (days 2-6), cytarabine 2 g/m2/day (days 2-6), filgrastim 300 μg/ day(days 1-6), and mitoxantrone 10 mg/m2/day (days 2-4)].
A repeat bone marrow biopsy was negative and PET scan showed near-complete resolution of the recurrent disease, with the periaortic nodule now measuring 4 mm with an SUV max of 2.7. Shethen underwent matched sibling donor allogeneic HSCT in November 2015with a myeloablative conditioning regimen of cyclophosphamide and total-body irradiation (dose 1,320 cGy).Cyclosporine and minidose methotrexate were used for graft-versus-host disease (GVHD) prophylaxis. There was no evidence of GVHD post-transplantation. At day +100, her bone marrow was negative for relapsed disease with 100% donor chimerism. Unfortunately, day +100 PET scan demonstrated FDG-avid anterior mediastinal, right hilar and left supraclavicular adenopathy, and CT-guided biopsy confirmed recurrent disease (Figure 2).
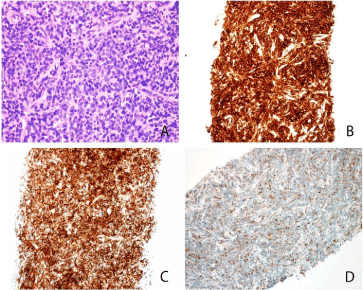
Figure 2: Histologic sections of the lymph node core biopsy show diffuse
involvement by an immature mononuclear cell infiltrate composed of
intermediate-sized cells with irregular nuclei, dispersed chromatin, and
variably prominent nucleoli (A). These immature cells are positive for CD33
(B), CD43 (C), and CD68/KP1(D); while negative for CD34, and B- and
T-lymphoid associated markers.
Shethen underwent radiation therapy to the mediastinal mass and began salvage treatment with the hypomethylating agent 5-azacitidine (32 mg/m2/day subcutaneous injection for 5 days, every 28 days), in lieu of more aggressive chemotherapy because of concerns for toxicity [2]. Tissue from a mediastinal lymph node biopsy was analyzed through a commercial next generation sequencing (NGS) based assay, which included the complete coding DNA sequences of 406 genes, as well as selected introns of 31 genes involved in rearrangements (Foundation One?, Boston, MA).This assay revealed several potentially targetable mutations (Table 1). One in particular, a fusion of ETV6-ABL1, was of interest because it forms a Philadelphia-like chromosome abnormality, t(9;12)(q34;p13), which could be targeted with a tyrosine kinase inhibitor [12-22]. Therefore, she was started on dasatinib (20 mg daily), at a lower-than-usual dose because of concurrent posaconazole mediated cytochrome P450 3A4 inhibition, in combination with 5-azacitidine at the same dose [3].
![]()
Genomic alterations detected
Potential targeted therapy
ETV6-ABL1 fusion
Tyrosine kinase inhibitors (TKIs), including bosutinib, dasatinib, imatinib, nilotinib, and ponatinib
MLLT10-PICALM fusion
DOT1L inhibitor (EPZ-5676)
NOTCH1 (K2150fs*100)
Gamma-secretase inhibitor (BMS-906024)
Table 1: Commercial next-generation sequencing (NGS) based assay (Foundation One?) identified several actionable genomic lesions for our patient, listed here alongside potential targeted therapies.
![]()
Kinase Gene
TyrosineKinase Inhibitor (TKI)
ABL
Bosutinib, dasatinib, imatinib, nilotinib, ponatinib
AXL
BGB324
BTK
Ibrutinib
CRLF2
Ruxolitinib
CSK
Dasatinib
EGFR1
Dovitinib (TKI258), gefinitib
EPOR
Ruxolitinib
EPH
Dasatinib, nilotinib
FGFR
Ponitinib
FLT3
Gilteritinib (ASP2215), lestaurtinib, midostaurin, quizartinib (AC220), semaxanib, sorafenib, sunitinib, tandutinib, E6201, KW-2449
JAK2
Ruxolitinib, AT9283
KIT
Imatinib
LYN
Bafetinib
MEK
Selumetinib, E6201
PDGFRA
Imatinib, nilotinib
PDGFRB
Imatinib
SRC
Dasatinib, imatinib
SYK
Entospletinib, TAK659
VEGFR
Ponitinib
Table 2: Numerous mutations have been described in acute myeloid leukemia and acute lymphoblastic leukemia that can be targeted with tyrosine kinase inhibitors (TKIs), with varying levels of evidence and success in vitro and in humans. In this table, all the currently described kinase-activating lesions in AML and ALL are matched with corresponding TKIs that are either FDA-approved or currently in development.
Unfortunately, follow up PET scan two months later revealed an FDG-avid lesion in the liver (2.1x2 cm) with an SUV max of 5.1, along with a 9 mm density in the left cerebellum. A biopsy of the liver lesion demonstrated relapsed myeloid sarcoma, and a magnetic resonance image (MRI) of the brain confirmed the presence of a cerebellar mass. She elected to undergo palliative radiation therapy to both lesions, and discontinued treatment with dasatinib. Consideration was given to treat her with ipilimumab after a recent phase 1/1b trial suggested that it can be feasible patients with extramedullary AML who relapse after HSCT [4]. However, a few weeks later, she became short of breath in the evening while vacationing with her family. The next morning, she died suddenly after a futile resuscitation attempt.
Discussion
ETV6-ABL1 (previously referred to in the literature as TEL-ABL) is an exceedingly rare fusion in acute myeloid leukemia (AML), appearing in just a single case (1/1197) in a screening study, and in just 6 case reports in literature to date [5-9]. In acute lymphoblastic leukemia (ALL), ETV6-ABL1 is also uncommon, and has been estimated to occur in 0.17% of cases in children, and 0.38% of cases in adults [10]. ETV6-ABL1 is also rarely encountered in BCR-ABL1 negative atypical chronic myeloid leukemia (CML) [11]. ETV6-ABL1 is similar to BCR-ABL1, with respect to the fact that it represents a gene fusion with tyrosine kinase activity that is associated with several types of hematologic malignancies. Eosinophilia seems to be a hallmark of ETV6-ABL1 rearranged leukemia, and the presence of this fusion oncogene is usually associated with a poor prognosis [6,10].
Interestingly, in vitro studies have shown that the ETV6-ABL1 fusion is TKI sensitive [12-14]. Moreover, 7 case reports have demonstrated that ETV6-ABL1 positive a typical CML responds to TKI therapy [15-21]. In addition, TKI therapy has been reported to help maintain disease remission in two patients with ETV6-ABL1 positive ALL, including an 8-year-old girl treated with imatinib, and an 82-year-old man treated with dasatinib [14,22]. Hence, there is reason to believe that ETV6-ABL1 positive AML could respond to targeted kinase inhibition.
It is unclear at what point our patient developed the ETV6-ABL1 fusion, since cytogenetic studies before NGS never demonstrated a lesion involving 9q34. However, in many case reports of ETV6-ABL1 positive hematologic malignancies, this genomic lesion was not revealed with conventional cytogenetic testing. Hence, it is unclear if the ETV6-ABL1 evolved late in the disease course after relapse, or if it was present before HSCT. If it were present earlier, then performing NGS early in the disease course could have been of clinical benefit.
It appears that for our patient, dasatinib in combination with low-dose 5-azacitidine lead to transient stability, but ultimately the patient relapsed. There are many plausible reasons why, including the possibility that dasatinib was temporarily effective, but lead to the evolution of a dasatinib-resistant sub-population of leukemic cells. Or perhaps ETV6-ABL1 was never a driver mutation, and targeting it was irrelevant. It is also very possible that since this is an unprecedented example of ETV6-ABL1 positive myeloid sarcoma, the dasatinib might not have had the desired pharmacologic effect.
TKIs like dasatinib have emerged as powerful tools to augment traditional chemotherapy regimens in patients with acute leukemia. Importantly, recent detailed genomic profiling of diseases such as Philadelphia chromosome-like ALL have demonstrated that genomic alterations amenable to inhibition with TKIs are more common than it was previously thought [14]. Hence, it is reasonable to assume that genomic lesions amenable to inhibition with TKIs could be relatively common in other types of leukemia as well, including AML. It is conceivable that ETV6-ABL1, and many other similar kinaseactivating lesions are underreported in the literature for diseases like AML. We summarize currently known kinase-activating lesions in AML and ALL in Table 2, alongside TKIs that target these lesions that are both Food and Drug Administration-approved and currently in development.
Even though there was ultimately a sub-optimal outcome for our patient with relapsed myeloid sarcoma, these fusion oncogenes are nevertheless prime targets for augmented therapy. We hope that this report attests that in the era of genomic medicine, lesions like ETV6- ABL1could be identified early in the disease course for patients and guide therapeutic strategies in the pursuit of better outcomes.
References
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012; 366: 1079-1089.
- de Lima M, Giralt S, Thall PF, de Padua Silva L, Jones RB, Komanduri K, et al. Maintenance therapy with low-dose azacitidine after allogeneic hematopoietic stem cell transplantation for recurrent acute myelogenous leukemia or myelodysplastic syndrome: a dose and schedule finding study. Cancer. 2010; 116: 5420-5431.
- Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA, et al. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011; 117: e75-e87.
- Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N Engl J Med. 2016; 375: 143-153.
- O'Brien SG, Vieira SA, Connors S, Bown N, Chang J, Capdeville R, et al. Transient response to imatinib mesylate (STI571) in a patient with the ETV6-ABL t(9;12) translocation. Blood. 2002; 99: 3465-3467.
- La Starza R, Trubia M, Testoni N. Clonal eosinophils are a morphologic hallmark of ETV6/ABL1 positive acute myeloid leukemia. Haematologica. 2002; 87: 789-794.
- Golub TR, Goga A, Barker GF, Afar DE, McLaughlin J, Bohlander SK, et al. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol. 1996; 16: 4107-4116.
- Park J, Kim M, Lim J, · Kim Y, Han K, Kim JS, et al. Variant of ETV6/ABL1 gene is associated with leukemia phenotype. Acta Haematol. 2013; 129: 78-82.
- Tirado CA, Siangchin K, Shabsovich DS, Sharifian M, Schiller G. A novel three-way rearrangement involving ETV6 (12p13) and ABL1 (9q34) with an unknown partner on 3p25 resulting in a possible ETV6-ABL1 fusion in a patient with acute myeloid leukemia: a case report and a review of the literature. Biomark Res. 2016; 4: 16.
- Zaliova M, Moorman AV, Cazzaniga G, Stanulla M, Harvey RC, Roberts KG, et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica. 2016.
- De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Basinko A, De Braekeleer M. ETV6 fusion genes in hematological malignancies: a review. Leuk Res. 2012; 36: 945-961.
- Carroll M, Ohno-Jones S, Tamura S, Buchdunger E, Zimmermann J, Lydon NB, et al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997; 90: 4947-4952.
- Okuda K, Weisberg E, Gilliland DG, Griffin JD. ARG tyrosine kinase activity is inhibited by STI571. Blood. 2001; 97: 2440-2448.
- Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371: 1005-1015.
- Gancheva K, Virchis A, Howard-Reeves J, Nick CP Cross, Brazma D, Grace C, et al. Myeloproliferative neoplasm with ETV6-ABL1 fusion: a case report and literature review. Mol Cytogenet. 2013; 6: 39.
- Kawamata N, Dashti A, Lu D, Miller B, Koeffler HP, Schreck R, et al. Chronic phase of ETV6-ABL1 positive CML responds to imatinib. Genes Chromosomes Cancer. 2008; 47: 919-921.
- Perna F, Abdel-Wahab O, Levine RL, Jhanwar SC, Imada K, Nimer SD, et al. ETV6-ABL1-positive "chronic myeloid leukemia": clinical and molecular response to tyrosine kinase inhibition. Haematologica. 2011; 96: 342-343.
- Tirado CA, Sebastian S, Moore JO, Gong JZ, Goodman BK. Molecular and cytogenetic characterization of a novel rearrangement involving chromosomes 9, 12, and 17 resulting in ETV6 (TEL) and ABL fusion. Cancer Genet Cytogenet. 2005; 157: 74-77.
- Kelly JC, Shahbazi N, Scheerle J, Jahn J, Suchen S, Christacos NC, et al. Insertion (12;9)(p13;q34q34): a cryptic rearrangement involving ABL1/ETV6 fusion in a patient with Philadelphia-negative chronic myeloid leukemia. Cancer Genet Cytogenet. 2009; 192: 36-39.
- Barbouti A, Ahlgren T, Johansson B, Höglund M, Lassen C, Turesson I, et al. Clinical and genetic studies of ETV6/ABL1-positive chronic myeloid leukaemia in blast crisis treated with imatinib mesylate. Br J Haematol. 2003; 122: 85-93.
- Kakadia PM, Schmidmaier R, Volkl A, Schneider I, Huk N, Schneider S, et al. An ETV6-ABL1 fusion in a patient with chronic myeloproliferative neoplasm: Initial response to Imatinib followed by rapid transformation into ALL. Leuk Res Rep. 2016; 6: 50-54.
- Malone A, Langabeer S, O’Marcaigh A, Storey L, Bacon CL, Smith OP. A Doctor(s) dilemma: ETV6-ABL1 positive acute lymphoblastic leukaemia. Br J Haematol. 2010; 151: 101-102.