
Research Article
Austin J Microbiol. 2015; 1(2): 1007.
β- Hydroxyacyl-acyl Carrier Protein Dehydratase (FabZ) from Candidatus liberibacter asiaticum: Protein Characterization and Structural Modeling
Wang S1, GAO Z2, LI Y2, Jiang L1* and Dong YH2*
¹Ministry of Education Key Laboratory of Plant Biology, Huazhong Agricultural University, China
²Institute of High Energy Physics, Chinese Academy of Sciences, China
*Corresponding author: Jiang LI, Ministry of Education Key Laboratory of Plant Biology, Huazhong Agricultural University, Wuhan 430070, China Yuhui Dong, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
Received: May 10, 2015; Accepted: September 01, 2015; Published: September 10, 2015
Abstract
β- Hydroxyacyl-acyl carrier protein (ACP) dehydratase (FabZ) is necessary for bacterial fatty acid biosynthesis. This protein catalyzes an essential step in the FASII pathway and in dehydration of β-hydrozyacyl-ACP to trans-2- acyl-ACP. Inhibition of β-hydroxyacyl-ACP dehydratase blocks the growth and reproduction of Candidatus liberibacter. β- Hydroxyacyl-ACP dehydratase is an attractive target for developing novel antimicrobials. In this study, the gene encoding enoyl-ACP dehydratase (FabZ) in C. liberibacter was cloned, expressed, and subjected to crystal structure analysis by X-ray diffraction. The optimum crystallization conditions were achieved with“Hampton research”pool of 2% v/v Tacsimate pH 5.0, 0.1 M sodium citrate tribasic dihydrate, pH 5.6, 16% w/v polyethylene glycol 3350 at 20oC, at a protein concentration of 7 mg/ mL. Small-angle X-ray scattering results showed that the ClFabZ protein was hexameric in solution. Crystal data were obtained using proteins produced in culture under the same conditions as those used for L-Se-Met-substituted ClFabZ. The crystal was in the space group of a lattice whose center was P6222, and the unit-cell parameters were a = b = 75.346 Å, c = 353.236 Å, a = β = 90°, and γ = 120°. These data were collected at a resolution of 2.9 Å. Furthermore, the interaction of hydrogen bonds and salt bridges in the ClFabZ structure were elucidated. Our results revealed the spatial structure information of FabZ in C. liberibacter asiaticum, which can serve as a basis for designing type-selective drugs by targeting FabZ.
Keywords: Candidatus Liberibacter asiaticus; β-Hydroxyacyl acyl carrier protein (ACP) dehydratase; Crystal structures; Small-angle X-ray scattering (SAXS)
Abbreviation
ACP: Enoyl–acyl Carrier Protein; BSRF: Beijing Synchrotron Radiation Facility; FAS: Fatty Acid Synthesis; HLB: Citrus Huanglongbing; PCR: Polymerase Chain Reaction; IPTG: Isopropyl β-d-Thiogalactopyranoside; PMSF: Phenylmethanesulfonyl Fluoride; SAXS: Small-angle X-ray Scattering
Introduction
Citrus Huanglongbing (HLB) is a citrus disease that poses serious obstacles in the development of the citrus industry owing to its propensity to invade uninfected regions [1,2], HLB causes severe loss in the production of citrus species such as sweet orange, mandarin, lemon, and grapefruit, among others [3,4]. Infected plants and scions of an infected citrus may spread HLB to other areas. The pathogen is disseminated principally by the insect vector citrus psyllid [5]. The pathogen acts as a parasite and resides in an intracellular sieve tube, disrupting nutrient transport and, consequently, metabolic activity in plants. HLB is caused by Candidatus liberibacter, which is a gram-negative bacterium belonging to the Rhizobiaceae family. The word Candidatus means that this organism cannot be maintained in culture [6]. Liberibacters are detected through polymerase chain reaction (PCR)-based amplification of their 16S rRNA genes using specific primers [7]. There are no effective prevention and control measures for HLB. In order to limit the outbreak of HLB and control the damage of economically important citrus trees, development of novel antibacterial agents against C. liberibacter is highly important.
Fatty acid biosynthesis is extremely important in all living cells. In general, fatty acid biosynthesis is classified into two different pathways (i.e., FASI and FASII) because of the enzyme architecture involved. Synthases involved in FAS pathway are large multifunctional enzymes with multiple domains that catalyze various reactions of FAS. Such synthases are found in mammals and fungi [8-10]. In contrast, FAS in plant chloroplasts and bacteria belong to the FASII pathway, in which; individual enzymes catalyze each step of biosynthesis. These enzymes are always highly specific in different bacteria [11,12].
FabZ catalyzes an essential step in the FASII pathway and dehydration of β-hydrozyacyl-ACP to trans-2-acyl-ACP [13-15]. Growth and reproduction of C. liberibacter is blocked when FabZ is inhibited. Therefore, β-hydroxyacyl-ACP dehydratase is an important target for the development of novel anti-bacterial agents. Crystal structures of several FabZs have been investigated in other microorganisms (e.g., protozoa). Currently, crystal structures of FabZs from several different sources, namely, Escherichia coli [16,17]. Helicobacter pylori [14,18,19]. Plasmodium falciparum [20-22], Clostridium acetobutylicium (Zhu et al., 2009), Campylobacter jejuni [23], Pseudomonas aeruginosa [24], and Burkholderia thailandensis E264 (Kim et al., unpublished) are known.
As a key enzyme in FAS synthesis, FabZ is an important target for the identification of novel antimicrobial compounds. Not surprisingly, considerable attention is currently focused on the identification and biochemical characterization of FabZ. EcFabZ has been shown to efficiently catalyze dehydration of short-chain β-hydroxyacyl-ACPs and long-chain saturated and unsaturated β-hydroxyacyl-ACPs in Escherichia coli [25]. TgFabZ of Toxoplasma gondii is expressed in both tachyzoites and bradyzoites. Indirect immunofluorescence assays showed that the TgFabZ protein localizes to the apicoplast, and a purified recombinant TgFABZ protein was found to be soluble and active [26]. Enzymatic characterization has been performed for FabZs from Enterococcus faecalis (EfFabZ) [27,28]. Pseudomonas aeruginosa (PaFabZ) [24], and P. falciparum (PfFabZ) [21,22,29]. Furthermore, the catalytic and inhibitory mechanisms of FabZ in Helicobacter pylori have also been characterized [18]. Specifically, three flavonoids targeting FabZ from Helicobacter pylori were identified, and the association of their crystal structure with enzymatic inhibition was characterized [18]. In the relative enzyme of fatty acid biosynthesis, FabF of Clostridium acetobutylicium functionally replaced FabZ in E. coli in vivo and in vitro but was unable to replace FabA, a key dehydratase–isomerase of E. coli in unsaturated fatty acid biosynthesis in vivo. FabA was also found to lack isomerase activity in vitro (Zhu et al., 2009). Moynié et al. reported that trans-2-enoyl-ACP polymerized in a progressive manner, and FabA in Pseudomonas aeruginosa underwent a second reaction that involved isomerization of trans-2- enoyl fatty acid to cis-3-enoyl fatty acid [30]. Lee et al. showed that E. coli SGJS25 cells over expressing the FabZ gene produced a high percentage of unsaturated fatty acids (UFAs), which constitute 35% of the total long-chain fatty acids in cells [17].
Numerous studies exist on the structure of FabZ. The structural features and sequences of FabZ indicate the presence of a“hot dog fold”[13] and (Pidugu et al., 2008). P. aeruginosa FabZ exhibits a hexameric crystalline structure (trimer of dimers) with a His/Glu catalytic dyad located within a deep, narrow tunnel formed at a dimer interface [24], The crystal structures of FabZ in Helicobacter pylori (HpFabZ) and its complexes with two inhibitors have been determined [14], In the present study, PfFabZ was found to exist as a homodimer (d-PfFabZ) in the crystals, in contrast with the reported hexameric form (h-PfFabZ), which is a trimer of dimers crystallized under a different condition. A crystalline structure of dimeric FabZ in P. falciparum revealed conformational switching to active hexamers by peptide flips [21], Chen et al. assayed the structural and thermodynamic characteristics of HpFabZ and emodin was enzymatically inhibited to target HpFabZ [19]. Structural bases for the functional and inhibitory mechanisms of FabZ in P. falciparum have also been reported [20,22], The structure of Pseudomonas aeruginosa FabA, which is an isozyme of FabZ, and interactions of the active site with its substrate were analyzed. Moynié et al. determined the structure of FabA in a complex with small molecules (Moynié et al., 2012). These studies have provided a detailed understanding of FabZ in C. liberibacter asiaticus.
Currently, C. liberibacter asiaticus cannot be cultured in vitro, which is a serious impediment to the development of vaccines against HLB. The recent sequencing of the C. liberibacter asiaticus genome has offered some hope for development on this front [31]. The present study aimed to clone, express, and purify the ClFabZ protein and subsequently elucidate the crystal structure of ClFabZ. Results of this study provide important insights for future efforts to discover antibacterial compounds that can target this enzyme.
Material and Methods
Cloning of C. liberibacter asiaticum fabZ gene
PCR was used to amplify the ClFabZ gene from the genomic DNA (NC_012985) [31]. Of Ca L. asiaticus strain using the forward primer 5´-gctaggatccatgatcagtgatcttgcatgc-3´ and the reverse primer 5´-tatactcgagaggattgttacccatgcgactc-3´. The amplified fragment of the FabZ gene was cloned into the pET28at-plus expression vector, in which an N-terminal His6-tag followed by a cleavable tobacco etch virus (TEV) protease site was introduced upstream of the BamHI site. The vector was then transformed into E. coli (DH5a) cells. All of the chemicals used were of reagent grade or ultrapure quality from Sigma.
Protein expression and purification of ClFabZ
The ClFabZ gene was transformed into E. coli BL21 (DE3) and the cells were cultured in LB media supplemented with 0.05 mg/mL of kanamycin at 37°C. The culture was induced by adding 0.4 mM of isopropyl-β-D-thiogalactopyranoside (IPTG) when its OD600 reached 0.8 – 1.0. The cultures were then incubated at 16°C for 20 h. The cells were harvested by centrifugation at 8000 rpm for 10 min at 4oC and later suspended in buffer A (20 mM Tris-HCl, pH 8.5, 500 mM NaCl, 1 mM PMSF). The cells were crash treated on ice, and the mixture yielded a clear supernatant by centrifugation at 16000 rpm for 60 min at 4°C. His-tagged FabZ was subsequently purified on a Ni–NTA agarose column using an imidazole gradient. The ClFabZ was eluted using 500 mM of imidazole. The resulting protein was digested by TEV protease, which was added to the protein at a 1:40 mass ratio, at 4oC overnight to remove the His6 tag and to obtain the native protein. The untagged protein was further purified in a Superdex 200 column (GE Healthcare) equilibrated with buffer B (20 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5 % (v/v) glycerol, 1mM DTT) using an AKTA purifier System (Amersham). Highly purified ClFabZ fractions were pooled and concentrated by ultrafiltration in an Amicon cell (Millipore, California, USA) to 20 mg/mL. These fractions were then quantified using a protein assay kit (Bio-Rad, USA).
The selenomethionine-labeled ClFabZ was expressed in E. coli BL21 (DE3). M9 medium containing extra amino acids (L-Semethionine at 60 mg/L; lysine, threonine and plenylalanine at 100 mg/L, respectively; leucine, isoleucine and valine at 50 mg/L, respectively) was used during expression. Purification of the Se- ClFabZ was performed as mentioned above. Each purification step was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Protein crystallization and data collection
A crystal of ClFabZ was obtained by the sitting-drop technique using a mixture of 1 µL of ClFabZ (7 mg/mL) and 1 µL of 2 % v/v Tacsimate pH4.0, 0.1 M Sodium citrate tribasic dihydrate pH 5.8, 15 % w/v Polyethylene glycol 3350. This mixture was equilibrated over a 100 µL-reservoir solution at 20°C. The crystal exhibited a stick shape.
All of the X-ray diffraction data were obtained on a similar detector beamline 1W3B (Beijing Synchrotron Radiation Facility). The crystals of the SeClFabZ were isomorphous with ClFabZ. All of the crystals were flash-cooled and maintained at 100 K by a flow of cold N2 gas during data collection.
Structure determination and refinement
The crystal structure of SeClFabZ was determined by singlewavelength anomalous dispersion method complemented with direct method to break the phase ambiguity [27,32-34]. The crystal structure of ClFabZ was determined using MolRep from CCP4 (Collaborative Computational Project 1994); the structure of SeClFabZ was used as the searching model, and the obtained structures were refined using the CNS program [35]. The qualities of all the models were assessed using the PROCHECK program [36]. A summary of the data collected and final refinement statistics performed is listed in Table 1.
SAXS measurements and data processing
Synchrotron SAXS measurements were performed at the European Molecular Biology Laboratory (EMBL) on the storage ring DORIS III of the Deutsches Elektronen Synchrotron (DESY, Hamburg). The instrument used was an X33 beamline equipped with a robotic sample changer and a PILATUS detector (DECTRIS, Switzerland). Scattering was recorded in the range of momentum transfer 0.07 < s < 5.5 nm-1, where s = (4πsinθ)/λ, 2θ is the scattering angle, and λ = 0.15 nm is the X-ray wavelength. Measurements were performed in a vacuum cuvette with an exposure time of 2 min to diminish parasitic scattering. The experimental scattering profiles from the samples were corrected for background scattering from the solvent. These profiles were then processed using standard procedures and the PRIMUS program.
The PRIMUS program was used to process the scattering curves [37]. The sample was measured at concentrations of 1, 2, and 4 mg mL-1 to exclude any concentration-dependent effects. The distance distribution function p(r) was computed using experimental data by the gnom program [38]. The theoretical curves were calculated using the crysol program [39,40].
Results and Discussion
Purification and oligomeric state of ClFabZ protein in solution
In this study, the FabZ gene from C. liberibacter asiaticus strain psy62 (ClFabZ) was cloned, and the recombinant expression plasmid pET28at-plus-ClFabZ was transformed into E. coli BL21 (DE3). The ClFabZ gene was a 486 bp fragment and included the stop codon. ClFabZ could be stably expressed in the E. coli system and purified using an Ni–NTA resin affinity chromatograph and highresolution gel filtration column (Superdex 200) following SDS-PAGE purification protocols. ClFabZ is a protein of 162 amino acids with a theoretical molecular mass of 18 kDa (Figure 1a). Interestingly, the chromatogram obtained from high-resolution gel filtration shows a single peak with an elution volume of 70 mL in the 120 mL-Superdex 200 column. The size of the protein that corresponds to a molecular mass of 119.61 kDa was investigated according to the linear regression equation of standard protein. This method was performed in a 120 mL-gel filtration column on Superdex G200. This finding indicated that ClFabZ was hexameric in solution. Organization of proteins in solution and in crystals may vary significantly because of the special conditions used during crystallization, which differ noticeably from physiological conditions. Therefore, the solution conformation of the protein was further studied using small-angle X-ray scattering (SAXS). The results were then compared with those obtained from crystal data. The trimeric and hexameric theoretical curves were compared with the experimental curve obtained from SAXS using crysol. The experimental data exhibited better fit with the hexameric theoretical curve (χ2 = 1.720) than the trimeric theoretical curve (χ2 = 8.529). These results revealed that CIFabZ formed a stable hexamer in solution (Figures. 1a–c).
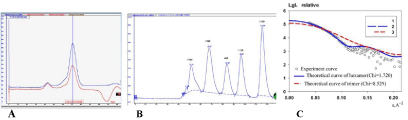
Figure 1: Detection of molar mass of ClFabZ using a Superdex G200 column and SAXS, (a): Analysis of ClFabZ by size-exclusion chromatography; (b): standard
protein analysis; and (c): fitting of theoretical scattering of trimers and hexamers to the experimental SAXS data. The experimental SAXS data are shown as balls,
while the theoretical scattering curve of trimers and hexamers are shown in blue and red lines, respectively.
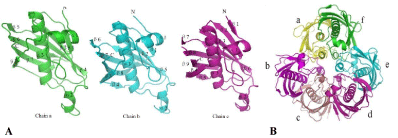
Figure 2: Structure of ClFabZ.
(A) Ribbon diagram of ClFabZ monomeric structure, where the secondary structural elements labeled N and C denote the N and C terminals of ClFabZ, respectively.
The parallel ß-sheet wraps around the central helix [a3] and (B) ribbon diagram of ClFabZ hexamer formed by two asymmetric units (chains a + b + c and chains
d + e + f) of the trimer. The view is from the three-fold axis of the hexamer. The dimer a/f is colored yellowish/green, the dimer b/c is colored rose/brick red, and
dimer e/d is colored cyan/peachblow.
Structure of apo-ClFabZ
The crystal data was obtained from proteins produced in culture under the same conditions as those used for L-Se-Met-substituted ClFabZ. X-ray diffraction data with 2.9 Å resolution was obtained. The crystal of the L-Se-Met-substituted ClFabZ belonged to the space group P6122 with unit-cell parameters of a = b = 75.346 Å, c = 353.236 Å, a = β = 90o, and γ = 120o. Table 1 shows the statistical results.
Structural features of ClFabZ
Each subunit of FabZ adopts a typical „hot dog“ fold, where six anti-parallel b-sheets wrap around a long central a-helix [13,41,42]. The dimer of hot dogs is formed by the association of the six ß-sheets from each subunit to form a continuous 12-stranded ß-sheet with central helices running anti-parallel to each other in H. pylori, P. aeruginosa, P. Falciparum, and B. thailandensis. However, the CjfabZ of C. Jejuni contain N-terminal ß-sheets and seven parallel ß-sheets that wrap around a long central a-helix. The crystal structure of ClFabZ in C. liberibacter shows an almost similar structure compared with the previously reported hexameric structure; the root mean square deviation (rmsd) of the Ca atoms of the hexamer was around 0.016 Å. However, three subunits, namely, the chains a, b, and c constituted an asymmetric unit of protein, and chains d, e, and f formed the other asymmetric unit. Two trimeric structures formed a hexamer, and the dimer of hot dogs was formed because of the association of the seven ß-sheets from each subunit. This association formed a continuous 14-stranded ß-sheet with the central helices that ran anti-parallel to each other in chain“a + f”of ClFabZ and eight ß-sheets and nine ß-sheets from each subunit to form a continuous 17-stranded ß-sheet with the central helices running anti-parallel to each other in chains“b + c”and“d + e”of ClFabZ, respectively. The whole structure may be considered a prism with three dimers as its sides (Figures 2A and 2B). When ClFabZ dimerized, two active site tunnels were formed at the dimer interface. These tunnels were long and cabined. Both of the subunits contributed to the formation of each tunnel. The exact mode of substrate binding was not clear because the loop that significantly contributed to stabilization of the substrate-binding tunnel was highly disordered in ClFabZ.
The interaction residues of ClFabZ
The H bonds and salt bridge were the main links between the asymmetric units of ClFabZ. The H bonds between the asymmetric units ABC–DEF refer to one H2O molecular link to both Y23 residues in chains c and d; one H2O molecular link to three residues: Y26-O, D29-OD1, K44-NZ in chain c and two residues T47-OG1, N49-N in chain d; and one H2O molecular link to three residues D-L26-O, D29-OD1, K44-NZ in chain d, and two residues T47-OG1, N49-N in chain c. The salt bridges K99-NZ-OD2-D98 produced between chains b and c and K99-N-OD2-D98 produced between chains a and f (Supplementary Table 1-1 & 1-2). The dimer formed was buried approximately1367 Å2 with extensive H bonds at the dimer interface (i.e., 17 pairs of H bonds, involving 11 kinds of residues, namely, Asp29, Lys30, Lys44, Asn45, Val46, Thr47, Phe48, Asp49, Glu50, Gly109, and Asp110). The same hydrogen bonds formed were involved in the dimers AF–ED and AF–BC. This finding was attributed to the symmetric structure observed (Supplementary Table 1-1& 1-2) and to hydrophobic interactions that stabilize the dimer structure. A total of 11 H bonds were formed between the monomeric chains b and c involved in 10 kinds of residues, namely, R22, G55, Y93, P105, M95, H102, I97, A100, P105, and P93. The salt bridges K99-NZ-OD2-D98 and K99-N-OD2-D98 were produced between the monomeric chains b and c (Supplementary Table 1-1 & 1-2), and the same H bonds and salt bridges were produced between the monomeric chains d and e (not shown).
Structural comparison of ClFabI with other ENR structures
Topology of the ClFabZ monomer was similar to those of other ENRs from different species. The results of structure-based Dali search [43] showed several hundreds of homologous structures. In the present study, the homologous structures from the top six species were selected and compared with ClFabZ. These species were Burkholderia thailandensis E264 (PDB ID: 4PIB, Kim et al., unpublished), Escherichia coli (GI:68304070), Pseudomonas aeruginosa (PDB ID: 1MKA; [24], P. falciparum (PDB ID:1Z6B, [20], Campylobacter jejuni (PDB ID:3D6X, [23], and Helicobacter pylori (PDB ID:2GLL, [14], and PDB ID:3ED0, [19].
Figure 3 shows the sequence alignment of ClFabZ with other FabZs from several other model organisms by DNAman. ClFabZ was found to share 38.8%, 35.6%, 40.9%, 33.5%, 41.8%, and 41.3% amino acid sequence identities with the homologous structures of B. thailandensis, E. coli, P. aeruginosa, P. falciparum, C. jejuni, and H. Pylori. The RMSD values varied from 0.016 Å to 1.565 Å when the apo-ClFabI was superposed to these apo structures. This result indicates that the structures were similar to one another, and some fragments at various locations were present (Figure 3). Two short a-helices formed at the N terminus, with one positioned at the beginning of the N terminus and the other between ß2 and a3 in ClFabZ. In ClFabZ, chain a was composed of a1/ß1/ß2/a2/a3/ß3/ß4/ ß5/ß6/ß7 while chain b was composed of a1/ß1/ß2/a2/a3/ß3/ß4/ß5/ ß6/ß7/ß8. The secondary structural elements were represented bate bridge before the a helix in chain c of ClFabZ: ß1/a1/ß2/a2/a3/ß3/ ß4/ß5/ß6/ß7/ß8/ß9, and the secondary structure percentage of the a-helix, ß-strand, and coil were 20%, 42% to 45%, and 38% to 35%, respectively. Interestingly, the structure of ClFabZ was found to be different from the structures of other bacterial FabZs. For instance, HpFabZ contains an extra short two-turn a-helices (a4) between a3 and ß3, which plays an important role in shaping a substrate-binding tunnel [18].
Figure 3: Structure-based sequence alignment of FabZ. Multiple sequence
alignment of FabZs from
H. pylori, C. jejuni, P. aeruginosa, P. falciparum, E.coli, and B.thailandensis.
Difference in lengths and composition of the sequence in different organisms
(highlighted by green box) leads to different structures. The totally conserved
residues that are highlighted by deep blue box share 100% homology. The
residues involved in the formation of hexamer (trimer of dimers) are marked
with purple squares. The conserved catalytic residues, namely, His and Glu,
are marked with orange squares. Alignment was performed using DNAman
software.
The phylogenetic tree of multiple sequence alignments of FabZ indicated that the sequence of ClFabZ approached that of C. Jejuni and HpFabZ (Figure 4). Five key motifs (i.e., PHRYPFLLVD, GHFP, PGVL, EALAQ, and PGD) were found to be conserved, with the exceptions of presence of Ile instead of Phe in BtFabZ, Met instead of Phe in HpFabZ, Met instead of Leu in EcFabZ and PaFabZ, and GM instead of AL in CjFabZ, HpFabZ and ClFabZ.
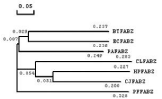
Figure 1: Phylogenetic tree of multiple sequence alignment of FabZ from
H. pylori, C. jejuni, P. aeruginosa, P. falciparum, C. liberibacter and B.
thailandensis. Data shown in the figure are branch length.
Analysis and prediction of active sites in ClFabZ
Alignment of ClFabZ and HpFabZ showed that their monomeric structures were similar, with the exception of the two short ß-sheets in ClFabZ that substitute one long ß-sheet in HpFabZ (Figure 5a). Moreover, alignment of ClFabZ and CjFabZ also showed that their monomeric structures were similar, with the exception of two short ß-sheets in ClFabZ that substituted one long ß-sheet in CjFabZ and one more a4 involved in CjFabZ (Figure 5b).
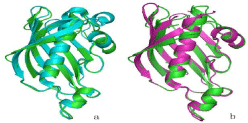
Figure 5: Overall survival, autologous stem cell transplant (ASCT) versus no ASCT (p=0.12).
Enzymatic activity assay of the HpFabZ mutant (Y100A) confirmed the importance of Tyr-100 in catalytic activity and substrate binding compared with ClFabZ. The residue Phe-83 at the exit tunnel was refined in two alternative conformations that caused the tunnel to form an L shape and a U shape [18]. The active site in HpFabZ was mainly hydrophobic and appeared as an L-shaped tunnel. The catalytically important amino acids His133 and Glu1470 (from the other subunit), together with His98, formed the only hydrophilic site in this tunnel. The inner end of the active site tunnel was closed by the phenyl ring of Phe169, which is located in a flexible, partly visible loop [22].
In this complex structure, the HpFabZ hexamer displayed a classical“trimer of dimers”organization similar to the native HpFabZ structure (PDB code 2GLL). A dimer was formed through hydrophobic interactions after every two monomers. Two L-shaped substrate-binding tunnels were located at the interface of the dimer. The entrance of these tunnels was protected by a door residue (Tyr100). The Tyr100 residue adopted two different conformations: an open conformation, in which the side chain of Tyr100 pointed towards Ile64’, allowed the chains of the substrates to enter the tunnel; a closed conformation hindered the substrate chain from reaching the catalytic site. The catalytic site in the tunnel was formed by two highly conserved residues, namely, His58 and Glu72’, that were located in the middle kink of the tunnel. Emodin connected the Tyr100 active site and prevented the substrate from integrating with HyFabZ [19].
The active sites of ClFabZ were defined by analyzing other known FabZ structures. These active sites may reside on the subunit–subunit interface, with each dimer containing two active sites. The catalyzed active site may be the conserved His57 and Glu71’ (indicating residue from the other subunits of the dimer) residues based on our analysis of other known FabZ structures, and may reside in the middle of the tunnel (Figure 6).
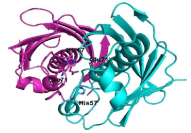
Figure 6: Prediction of the active site of ClFab.
Conclusion
In this study, FabZ was investigated as an important target for antibacterial drug design because FabZ is a potent enzyme in bacterial FAS. Furthermore, this enzyme catalyzes the dehydration of ß-hydroxyacyl-ACP to trans-2-acyl-ACP in bacteria. Sequence alignment results showed that ClFabZ belongs to a conserved gene family. ClFabZ forms a hexamer in its native state, as observed in SDSPAGE and SAXS results. Its high stability may be ascribed to strong hydrophobic interactions and strong H bonds among its hexamers, as evidenced by our structural modeling data. The structural features and sequences of proteins that exhibited a“hot dog fold”were further analyzed in this study.
This study reveals that significant differences exist in the protein sequences of FabZ although the basic architecture of the fold is well conserved in these proteins. Segments with certain conserved sequence motifs play crucial structural and functional roles in various classes of these proteins. Based on secondary structure prediction using the ExPASy server, a loop composed of 12 amino-acid residues in the N-terminus may likely influence its crystallization. Moreover, some of these residues were not found to be conserved, and their function has not been characterized to date. Thus, the present study offers novel insights into FabZ of the C. liberibacter asiaticus strain and provides a foundation for the discovery of anti-bacterial compounds using ClFabZ as the target.
Acknowledgment
This work was supported by grants from the National Basic Research Program of China (2012CB917203) and the National Natural Science Foundation of China (Nos. 31272146, 10979005, and 31100531). The authors are grateful for the assistance provided by Dr. Wenjia Wang in analysis with SAXS.
References
- Bove JM. Huanglongbing: A destructive, newly emerging, century-old disease of citrus. J. Plant Pathol. 2006; 88: 7-37.
- Davi RI and Ruabete TK. Records of plant pathogenic viruses and virus-like agents from 22 Pacific island countries and territories: a review and an update. Australasian Plant Pathology. 2010; 39: 265-291.
- Halbert SE, Manjunath KL. Asian citrus psyllids (Sternorryncha:Psyllidae) and greening disease of citrus:a literature review and assessment of risk in Florida. Fla. Entomol. 2004; 87: 330-353.
- Coletta-Filho HD, Takita MA, Targon MLPN, Machado MA. Analysis of 16S rDNA sequences from Citrus Huanglongbing bacteria reveal a different“Ca. Liberibacter”isolate associated with Citrus disease in Sao Paulo. Plant Dis. 2005; 89: 848-852.
- Halbert SE. The discovery of huanglongbing in Florida. In: Proceedings of the 2nd international citrus canker and Huanglongbing research workshop: Florida Citrus Mutual, Orlando. 2005.
- Jagoueix S, Bove JM, Garnier M. The phloem-limited bacterium of greening disease of citrus is a member of the alpha subdivision of the Proteobacteria, Int. J Syst Bacteriol. 1994; 44: 379-386.
- Li W, Hartung JS, Levy L. Quantitative real-time PCR for detection and identification of Candidatus Liberibacterspecies associated with citrus huanglongbing. Journal of microbiological methods, 2006; 66: 104-115.
- Maier T, Jenni S, Ban N. Architecture of mammalian fatty acid synthase at 4.5? resolution. Science. 2006; 311: 1258-1262.
- Kiema TR, Engel CK, Schmitz W, Filppula SA, Wierenga RK, Hiltunen JK. Mutagenic and enzymological studies of the hydratase and isomerase activities of 2-enoyl-CoA hydratase-1, Biochemistry. 1999; 38: 2991-2999.
- Hiltunen JK, Wenzel B, Beyer A, Erdmann R, Fossa A, Kunau WH. Peroxisomal multifunctional beta-oxidation protein of Saccharomyces cerevisiae. Molecular analysis of the fox2 gene and gene product. J Biol Chem.1992; 267: 6646-6653.
- Tasdemir D. Type II fatty acid biosynthesis, a new approach in antimalarial natural product discovery. Phytochemistry Reviews. 2006; 5: 99-108.
- Döormann P, Synthesis L. The structure and function of plastids advances in photosynthesis and respiration. Metabolism and Transport. 2006; 23: 335-353.
- Dillon SC, Bateman A. The Hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinformatics. 2004; 5: 109.
- Liu WZ, Luo C, Han C, Peng SY, Yang YM, Yue JM, et al. A new b-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Helicobacter pylori: Molecular cloning, enzymatic characterization, and structural modeling. Biochemical and Biophysical Research Communications. 2005; 333: 1078-1086.
- Surolia A, Modak R, Sharma S, Ramya NSC, Kumar S, Kumar G, et al. Dehydratase (FabZ) -Hydroxyacyl-Acyl Carrier protein ß-Plasmodium falciparum inhibition of identification, characterization and enzyme catalysis and regulation. J. Biol. Chem.2003; 278: 45661-45671.
- Siddiqui R, Malik H, Sagheer M, Jung Suk-Yul, Khan NA. The type III secretion system is involved in Escherichia coli K1 interactions with acanthamoeba. Experimental Parasitology. 2011; 128: 409-413.
- Lee Sunhee, Jung Yeontae, Lee Seunghan, Lee Jinwon. Correlations between FAS Elongation Cycle Genes Expression and Fatty Acid Production for Improvement of Long-Chain Fatty Acids in Escherichia coli. Appl Biochem Biotechnol. 2013; 169: 1606-1619.
- Zhang L, Liu WZ, Hu TC, Li Du, Luo C, Chen KX, et al. Inhibitory Mechanisms of ß-Dehydratase (FabZ) Hydroxyacyl-acyl Carrier Protein. J Biol Chem. 2008; 283: 5370-5379.
- Chen J, Zhang L, Zhang Y, Zhang H, Du J, Ding J, et al. Emodin targets the beta-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Microbial. 2009; 9: 91-91.
- Maity K, Venkata BS, Kapoor N, Surolia N, Surolia A, Suguna K. Structural basis for the functional and inhibitory mechanisms of b-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Plasmodium falciparum. Journal of Structural Biology. 2011; 176: 238-249.
- Swarnamukhi PL, Sharma SK, Bajaj P, Surolia N, Surolia A and Suguna K. Crystal structure of dimeric FabZ of Plasmodium falciparum reveals conformational switching to active hexamers by peptide flips. FEBS Lett.2006; 580: 2653-2660.
- Kostrewa D, Winkler FK, Folkers G, Scapozza L, and Perozzo R. The crystal structure of PfFabZ, the unique ß-hydroxyacyl-ACP dehydratase involved in fatty acid biosynthesis of Plasmodium falciparum. Protein Science.2005; 14: 1570-1580.
- Kirkpatrick AS, Yokoyama T, Choi KJ, Yeo HJ. Campylobacter jejuni fatty acid synthase II: structural and functional analysis of beta-hydroxyacyl-ACP dehydratase (FabZ). Biochem Biophys Res Commun. 2009; 380: 407-412.
- Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, et al. The Structure of (3R)-Hydroxyacyl-Acyl Carrier Protein Dehydratase (FabZ) from Pseudomonas aeruginosa. J Biol Chem.2004; 279: 52593-52602.
- Heath RJ, Rock. CO.Roles of the FabA and FabZ ß-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J Biol Chem. 1996; 271: 27795-27801.
- Dautu G, Ueno A, Munyaka B, Carmen G, Makino S, Kobayashi Y, et al. Molecular and biochemical characterization of Toxoplasma gondii ß-hydroxyacyl-acyl carrier protein dehydratase (FABZ). Parasitol Res. 2008; 102: 1301-1309.
- Wang H, Cronan JE. Functional Replacement of the FabA and FabB Proteins of Escherichia coli Fatty Acid Synthesis by Enterococcus faecalis FabZ and FabF Homologues. J Biol Chem.2004; 279: 34489-34495.
- Lu YJ, White SW, Rock CO. Domain Swapping between Enterococcus faecalis FabN and FabZ proteins localizes the structural determinants for isomerase activity. J Biol Chem.2005; 280: 30342-30348.
- Sharma SK, Kapoor M, Ramya TN, Kumar S, Kumar G, Modak R,et al. Identification, characterization and inhibition of Plasmodium falciparum beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J Biol Chem. 2003; 278: 45661-45671.
- Moynié L, Leckie SM, McMaho SA, Duthie FG, Koehnke A, Taylor JW, et al. Structural insights into the mechanism and inhibition of the ß-Hydroxydecanoyl-Acyl carrier protein dehydratase from Pseudomonas aeruginosa. J Mol Biol. 2013; 425: 365-377.
- Duan Y, Zhou L, Hall DG, Li W, Doddapaneni H, Lin H,et al. Complete genome sequence of citrus huanglongbing bacterium ‘Candidatus Liberibacter asiaticus' obtained through metagenomics. Mol. Plant Microbe Interact. 2009; 22: 1011-1020.
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr.1999; 55: 849-861.
- Cohen SX, Jelloul MB, Long F, Vagin A, Knipscheer P, Lebbink J, et al. ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr. 2001; 57: 1445-1450.
- Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003; 59: 38-44.
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination, Acta Cryst. 1998; 54: 905-921.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereo chemical quality of protein structures. J Appl Crystallogr. 1993; 26: 283-291.
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003; 36: 1277-1282.
- Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992; 25: 495-503.
- Svergun DI, Barberato C, Koch MHJ. CRYSOL-a program to evaluate X-ray scattering of biological macromolecules from atomic coodinates. J Appl Crystallogr.1995; 28: 768-773.
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001; 80: 2946-2953.
- Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure. 1996; 4: 253-264.
- Pidugu LS, Maity K, Ramaswamy K, Surolia N, Suguna K. Analysis of proteins with the 'hot dog' fold: Prediction of function and identification of catalytic residues of hypothetical proteins. BMC Structural Biology. 2009; 9: 37.
- Holm L, and Rosenström PD. Server: conservation mapping in 3D. Nucl. Acids Res. 2010; 38: W545-549.