
Review Article
Austin J Mult Scler & Neuroimmunol. 2015;2(1): 1009.
Targeting Inflammatory T Cells in Multiple Sclerosis: Current Therapies and Future Challenges
Loretta Tuosto*
Istituto Pasteur-Fondazione Cenci Bolognetti,Department of Biology and Biotechnology “Charles Darwin”, Sapienza University, Rome, Italy
*Corresponding author: Loretta Tuosto, Department of Biology and Biotechnology “Charles Darwin”, Sapienza University, 00185-Rome, Italy
Received: September 15, 2014; Accepted: February 18, 2015; Published: February 20, 2015
Abstract
Multiple Sclerosis (MS) is an autoimmune inflammatory disorder of the Central Nervous System (CNS), affecting more than one million people worldwide. The pathogenesis of MS involves several genetic and environmental factors, which ultimately lead to the activation of autoreactive T cells in the periphery, their migration into the CNS, where they trigger an acute inflammatory response, thus mediating primary demyelination and axonal damage. Most information on MS derives from studies in animal models of experimental autoimmune encephalomyelitis (EAE), which exhibit many similarities to the pathology of MS. Two distinct subsets of autoreactive T cells have been primarily involved in the pathogenesis of both EAE and MS: the interferon (IFN)-γ producing CD4+ T helper (Th) 1 and interleukin (IL)-17 producing Th17 cells. The activity of these cells is controlled by specific regulatory T cells (Treg), which by secreting antiinflammatory cytokines such as IL-4, IL-10 and tumour growth factor (TGF)-β efficiently inhibit Th1 and Th17 cells.
In this review, we summarize current knowledge on the role and function of pro-inflammatory and Treg subsets in MS. We also discuss the action of current and novel therapies aimed to dampen inflammatory T cells.
Keywords: Multiple sclerosis; Inflammatory T cells; Regulatory T cells;Therapy; Costimulation
Abbreviations
MS: Multiple Sclerosis; CNS: Central Nervous System; EAE: Experimental Autoimmune Encephalomyelitis; IFN: Interferon; Th: T helper; IL: Interleukin; TGF: Tumour Growth Factor; RR: Relapseremitting; SP: Secondary Progressive; PP: Primary Progressive; BBB: Blood-Brain Barrier; APC: Antigen Presenting Cell; DC: Dendritic Cell; MBP: Myelin Basic Protein; PLP: Proteolipid Protein; MOG: Myelin Oligodendrocyte Glycoprotein; Tregs: Regulatory T Cells; GA: Glatiramer Acetate; GITRL: Glucocorticoid-induced TNF Receptor Ligand; ASEs: Adverse Side-Effects; NK: Natural Killer; TCR: T Cell Receptor; PI3K: Phosphatidylinositol-3 Kinase; PIP2: Phosphatidylinositol 3,4-biphosphate; PIP3: Phosphatidylinositol 3,4,5-triphosphate; Ab: Antibody; GITR: Glucocorticoid-Induced TNF Receptor-related Protein
Introduction
MS is an autoimmune chronic inflammatory disorder characterized by demyelination and remyelination events and by the loss of sensory and motor functions. Two third of MS patients present the relapsing-remitting (RR) course, which is characterized by relapses usually followed by periods of recovery or remission, but one third of patients progresses to chronic secondary progressive (SP) disease [1,2]. A minority of patients (10-20%) experiences a primary progressive disease (PP), which is characterized by a gradual and constant decline in their neurological functions from the onset of disease [3]. Inflammation is present at all stages of MS [4] and pro-inflammatory cytokines/chemokines play a critical role in the pathophysiology of MS by compromising the blood- brain barrier (BBB) integrity, recruiting immune cells from the periphery, and activating resident microglia. Conversion of MS from RR to progressive phases has been related to prolonged chronic inflammation in the CNS. Moreover, both SPMS and PPMS patients have generalized inflammation in the whole brain accompanied by cortical demyelination and diffuse white matter injury [4].
Although several cell types within the CNS may contribute to the production of pro-inflammatory cytokines and chemokines, activated autoreactive T cells have a key role in inflammatory demyelination [5]. Indeed, the cytokine and chemokine-producing phenotype of self-reactive T cells in MS patients determines the ability of these cells to cross BBB and cause inflammation in the CNS, thus contributing to disease progression.
This article reviews the current knowledge on the contribution of different T cell subsets in the pathogenesis of MS and discusses the current and novel therapeutic strategies, which aim to dampen the pathogenic inflammatory T-cell response.
Inflammatory T helper cell subsets in MS: Th1 and Th17 cells
The more accepted pathogenic model for both EAE and MS is that autoreactive myelin-specific CD4+ T cells are activated in the periphery, entered the CNS by crossing the BBB and are reactivated by resident antigen-presenting cells (APC), mainly of which is microglial cells [5,6]. The priming and activation of autoreactive myelin-specific CD4+ T cells likely occurs in peripheral lymph nodes, where the dendritic cells (DC) may present myelin epitopes to naive T cells, thus inducing the activation and differentiation of autoreactive effector/memory T helper cells, which in turn migrate to the CNS and cause tissue damage and demyelination. Several evidences support a function for myelin proteins, such as MBP (myelin basic protein), PLP (proteolipid protein) and MOG (myelin oligodendrocyte glycoprotein), as relevant antigens in both EAE and MS [7-11]. In EAE, myelin-specific T cell responses seem to initiate in the CNSdraining cervical lymph nodes, thus suggesting that myelin proteins are constitutively presents in some lymph nodes [12]. Moreover, high avidity myelin-specific CD4+ T cells have been isolated from the periphery of MS patients [13,14].
It has been thought for a long time that the pathogenetic cells mediating MS were CD4+ Th1 cells, producing large quantity of IFN-γ driven by IL-12 [15,16]. Indeed, IFN-γ-deficient mice as well as knockout mice for IL-12p40, the large subunit of IL-12, were resistant to EAE [17-20]. However, further observations that mice deficient in IL-12p35, the smaller subunit of IL-12, and IL-12Rβ2 were susceptible to EAE [21,22] as well as data showing that the administration of IL-12 during the early phases of EAE suppressed EAE in an IFN-γ dependent manner [23], suggested that Th1 cells could not be responsible for the pathogenesis of MS. The discovery that IL-23 shares the p40 subunit with IL-12 clarifies these contradictory data [24]. Cua et al. demonstrated that IL-23 rather than IL-12 is crucial for EAE development by showing that IL-23p19, the smaller subunit of IL-23, deficient mice were resistant to EAE [25,26]. Data from Langrish et al. clearly defined the role of IL-23 in favouring EAE by driving and inducing the expansion of a novel Th subset producing IL-17 [26], designated Th17 cells [27,28].
The hallmark of Th17 cells is the production of the proinflammatory cytokine IL-17 (A and F) that affects the functions of a wide range of cells, enhances the secretion of other pro-inflammatory cytokines and chemokines and increases the activation of matrix metalloproteinases, thus contributing to the breakdown of BBB [29]. In the human system, Th17 differentiation can be mediated by IL- 6, TGF-β and IL-21, while IL-23 is critical in maintaining the Th17 phenotype [30-35]. Several data demonstrated that CD4+ Th17 subset exerts a central role in the pathogenesis of both EAE and MS. Increased numbers of Th17 cells have been found in the both inflamed CNS [36] and in the periphery of acute EAE [5] as well as in the CSF of MS patients during relapses [37]. High levels of IL-17A have been also detected in circulating leukocytes of MS patients with active disease [38], and higher IL-17A production has been correlated with the number of active plaques on magnetic resonance imaging [39]. All these data strongly support a pivotal contribution of Th17 cells in the pathology of MS [40].
Regulatory T cells (Tregs)
Tregs are negative regulators of T helper cell responses and contribute to T cell “anergy” and to the maintenance of self- tolerance, thus protecting against autoimmunity [41,42]. Tregs can be identified through their surface phenotypes and cytokine producing profiles. Tregs may be divided into two main subsets, natural Tregs (nTregs) and inducible Tregs (iTregs). The CD4+CD25+ FOXP3+ nTregs develop in the thymus and their TCR repertoire is skewed towards the recognition of self-antigens. In contrast, iTregs, originate from naive T cells in the peripheral lymph nodes, are either CD4+ or CD8+ and may or not express FOXP3 [43].
Dysfunctions or impairment maturation of Tregs have been observed in animal models of MS [44]. The presence of myelinspecific Tregs within the CNS during EAE highlights their role in the control of disease. Indeed, the accumulation and frequency of Tregs in the CNS has consistently been shown to correlate with recovery from EAE. However, myelin-specific Tregs were not sufficient to reduce the function of encephalitogenic effector T cells during the peak of EAE [45].
In MS, several reports have shown that human Tregs are functionally impaired, have decreased FOXP3 expression compared to healthy individuals, or have deficit in their maturation, or in their thymic emigration [44]. Reduced number or impaired suppressive functions of Tregs have been also found in the peripheral blood of MS patients [46-49]. Therefore, dysfunction in the number and/or functions of Tregs may concur to the immunopathogenesis of MS by decreasing the suppression of activated pathogenic immune cells (Figure 1).
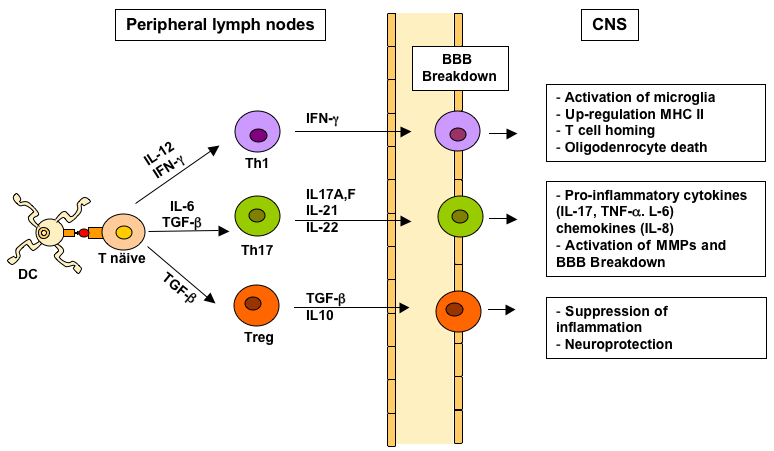
Figure 1: CD4+ Th cell subsets in MS. Naive CD4+ T cells are activated
in peripheral lymp nodes by myelin-like peptide presented on dendritic cells
(DC). Specific cytokines direct the polarization of activated Th cells into Th1
(IL-12, INF-γ), Th17 (IL-6 and TGF-β) of Treg cell subsets (TGF-β). Activated
Th cells leave the limph nodes and migrate into the CNS across the BBB.
In the CNS, Th cells interact with resident microglia, are reactivated upon
recognition of myelin antigens and produce cytokines and chemokines that
lead (Th1 and Th17) or suppress (treg) the inflammatory cascade in the CNS
and damage.
T-cell based therapeutic strategies
Our understanding of the key role of T cells in MS has led to a vast research during the last two decades in an attempt to develop novel therapeutic strategies for T-cell immunotherapy. Below are summarized some that are currently used and others that required further studies.
First-line treatments: IFN-β and glatiramer acetate (GA)
The most common disease modifying drugs used in the first-line treatment of RRMS include IFN-β or GA. Although the mechanisms of action of these compounds are still poorly understood, they exert some effects on T lymphocytes.
IFN-β belongs to a family of cytokines, which interfere with virus replication and exhibit immunomodulatory activities. In 1981, the administration of IFN-β to MS patients resulted in a significant reduction of the relapse rate [50], thus encouraging further clinical trials [51]. IFN-β is actually approved as first-line therapy for RRMS, SPMS with additional relapses in the European Union (EU) [52], but not for PPMS patients, who showed no significant reduction of disability after treatment [53].
The immunomodulatory activities of IFN-β in ameliorating RRMS include its ability to interfere with T cell activation by inhibiting the up-regulation of MHC class II molecules induced by IFN-γ on APC [54] as well as to inhibit costimulatory molecule functions (i.e. CD40/CD40L and CD28/B7) [55,56]. More recently, the immunosuppressive effects of IFN-β were related to immune deviation. Distinct reports showed that IFN-β treatment impairs Th17 cells by reducing IL-23 levels and up-regulating IL-10, which has general suppressive effects on Th cells or by inducing IL-27, which inhibits Th17 cell differentiation [57-59]. A role of IFN-β in inducing Tregs has also been evidenced [60,61]. In a longitudinal followup stadium performed on stable RRMS patients before and after 6-months of IFN-β therapy, Namdar et al. evidenced a significant increase of both frequency and suppressive functions of Treg cells of IFN-β- treated patients [61]. Increased frequency of Treg cells was also observed in EAE mice after IFN-β treatment. This event seems to be correlated to enhanced proliferation of Treg cells mediated by the up-regulation of glucocorticoid- induced TNF receptor ligand (GITRL) on the surface of dendritic cells [60].
Besides the beneficial effects of IFN-β in MS, common side effects have been reported, which include flu-like symptoms, injectionside reactions (i.e. pain, swelling, lypoatrophy), lymphocytopenia and elevated liver enzymes. Moreover, a significant proportion of MS patients (between 7 % to 49%) do not respond to IFN-β [62]. One of the main causes of IFN-β treatment failure is related to the development of neutralizing anti-IFN-β antibodies [63].
GA is a copolymer of alanine, lysine glutamic acid and tyrosine that exerts multiple immunomodulatory activities. Its immunosuppressive effects on Th cells has been related to the inhibition of IFN-γ and stimulation of IL-4 secretions, thus favouring a shift of the immune response from pro-inflammatory Th1 to anti-inflammatory Th2 [64,65]. Another mechanism of the anti-inflammatory effects of GA is the expansion of Tregs through the up-regulation of FOXP3 [66,67]. However, beneficial effects of GA administration were demonstrated only in RRMS [68,69].
In summary, the mechanisms of action of either IFN-β or GA partly target T-cell mediated inflammatory response in MS. However, both drugs often fail to exhibit sufficient clinical effectiveness and have very little effects in progressive phases of the disease that is currently treated with mitoxantrone, an immunosuppressive cytotoxic drug with immunomodulatory activity, but with serious side effects, such as myelosuppression and cardiac toxicity (Table 1) [70-72].
![]()
Drug
Mode of action
Approved in Europe
Major side-effects
IFN-?
Immunomodulation:
- Reduction of Il-23 and upregulation of IL-10 levels
- Expansion of Tregs
- RRMS (first-line)
- SPMS with relapses
- Flu-like symptoms
- Injection-side reactions
- Hepatotoxicity
Glatiramer acetate (GA)
Immunodulation:
- Reduction of IFN-γγ(Th1) and up-regulation of IL-4 (Th2)
- Expansion of Tregs
- RRMS (firs-line)
- Flu-like symptoms
- Injection-side reactions (lipoatrophy)
Dimethyl-fumarate
Immunomodulation:
- Depletion of peripheral blood T cells
- Reduction of IFN-γ(Th1) and up-regulation of IL-4 (Th2)
- RRMS (first-line)
- Flushing
- Gastrointestinal side effects
- Hepatotoxicity
- Leukopenia
Terifluoomide
Inhibition �of �proliferation of active dividing cells
- RRMS (first-line)
- Gastrointestinal side effects
- Hair loss
- Hepatotoxicity
- Leukopenia
- Teratogenic potential
Natalizumab
Blocking migration of immune cells to the CNS
- RRMS (second-line)
- Allergic infusion reactions
- Infections (PML)
- Hepatotoxicity
Fingolimod
Blocking egress of lymphocytes from lymph nodes
- RRMS (second-line)
- Infections (HSV, VZV)
- Bradycardia and arrythmias
- Macular edema
Alemtuzumab
Depletion of immune cells expressing CD52 (i.e. T cells)
- RRMS (highly active)
- Allergic infusion reactions
- Infections (HSV)
- Autoimmune disorders (thyroid and Good-pasture syndrome)
Mitoxantrone
Inhibition of proliferation of active dividing cells
- RRMS (highly active)
- SPMS
- Cardiotoxicity
- Gastrointestinal side effects
- Acute myeloid leukemia
PML: Progressive Multifocal Leukoencephalopathy; HSV: Herpes Simplex Virus; VZV: Varicella Zoster Virus
Table 1: Currently approved therapies in MS.
Second-line therapeutic drugs
Second-line drugs, commonly used in case of failure or intolerance to the first-line therapeutic agents, include natalizumab, fingolimod and cladribine. Generally, these agents are more potent than those used in the first-line therapy, but exhibit potential adverse side effects (ASEs) [73,74].
Natalizumab is a humanized recombinant monoclonal antibody directed against the a4 subunit of a4β1 and a4β7 integrins expressed on lymphocytes. In particular, the integrin a4b1 binds to the vascular-cell adhesion molecule (VCAM)-1 on endothelial cells of brain and spinal cord vessels, thus allowing immune cells to cross BBB. Natalizumab prevents the migration of lymphocytes into the CNS [75]. In humans, natalizumab strongly reduces the numbers of CD4+ T lymphocytes in the CNS [76]. The results obtained by both AFFIRM and SENTINEL clinical trials demonstrated a strong reduction of both the annualized relapse rate and disability progression in natalizumab-treated RRMS patients [77,78]. However, it may induce severe secondary effects [74,77,79] and clinical complications such as progressive multifocal leukoencephalopathy by polioma JC virus [80].
Fingolimod (FTY720) binds to sphingosine-1-phosphate receptors (S1PR) and blocks the egress of both T and B cells from secondary lymphoid organs into circulation. Moreover, due to its lipophilic properties, fingolimod may cross the BBB and by binding the S1PR expressed on astrocytes, oligodendrocytes and microglia, may promote their survival and favour remyelination [81,82]. The results obtained by both TRANSFORMS [83] and FREEDOMS [84] clinical trials showed a strong reduction in both relapses and accumulation of new or enlarging T2 lesions. Moreover, the availability of oral active forms of fingolimod may also circumvent the ASEs (e.g. skin indurations and lipoatrophy) related to infusion or injection and may exhibit higher adherence and long-term persistence [85]. However, important ASEs, including infections, arrhythmias, hypertension, macula edema as well as serious ASEs, such as asystole, bradycardia and malignant neoplasm have been reported [83,84,86,87]. More recently, an observational study by Carruthers et al. aimed to compare the effectiveness of natalizumab and fingolimod in clinical practice evidenced the superior effectiveness of natalizumab [88]. Thus, a deeper assessment of the effectiveness, safety and adherence during a long-term period of treatment should be required be required.
Cladribine is a synthetic deoxyadenosine analogue used as an oral immunomodulatory drug in the treatment of chronic progressive MS patients [89-91]. Cladibrine targets lymphocytes resulting in sustained reduction of peripheral T and B lymphocytes. The administration of oral cladribine resulted in reducing relapse-rates and disability progression in patients with RRMS [92-95]. Despite the strong lymphopenia observed in patients treated with oral cladibrine, no significant differences in the overall incidence of infections were observed. However, herpes zoster infections as well as neoplasm occurred in cladribine-treated patients, thus suggesting further monitoring to weigh the benefits against potential risks.
Other anti-inflammatory drugs are approved as second line-line therapy (Table 1). However, most of these non-specific therapeutic strategies compromise the ability of T cells to fight infections or to control cancer development and progression, thus more studies are required to determine the most efficacious, safe and tolerable treatment.
Targeting specific surface molecules or soluble cytokines for dampening inflammatory T cells in MS
Other therapies in clinical trials include the use of specific antibodies (Abs) targeting T cells or pro-inflammatory cytokines.
Daclizumab is a humanized monoclonal Ab directed against CD25. CD25 is the alpha chain of the high affinity IL-2 receptor. CD25 is up-regulated on activated T cells and is necessary for IL-2-driven survival and proliferation of activated T cells, including autoreactive cells. Binding of daclizumab to CD25 blocks the survival and proliferation of activated T cells through the preferential expansion of regulatory CD16-CD56bright natural killer (NK) cells with anti- viral and anti-tumour activities [96,97]. In phase III trials, daclizumab resulted in a significant reduction of the relapse rate and disability progression by more than 50% [98]. However, the ASEs reported in both phase II and III clinical trials were higher rate of infections, skin disorders, diffuse lymphadenopathy, and elevated liver enzymes [98- 100].
Alemtuzumab is a humanized monoclonal Ab against CD52, a surface glycoprotein expressed on several immune cells including T and B cells, eosinophils and monocytes. Its selective expression on mature cells but not on lymphoid progenitors made it a good target for immunotherapy [101]. The interaction of alemtuzumab with CD52 leads to the depletion of CD52+ cells [102], through complement and/ or cell mediated lysis [103]. Data from clinical trials evidenced a strong reduction of relapses and neurological disability in RRMS patients [104,105]. However, given the prolonged immunosuppression after treatment with alemtuzumab, its impressive effectiveness was associated with a high risk of thyroid autoimmune diseases [106]. Thus, the drug was not approved by the FDA due the negative risk/ benefit ratio but is currently approved in Europe (Table 1).
Despite the encouraging data obtained by the administration of neutralizing anti-IL-12 or anti-IL23p19 monoclonal Abs in EAE [107-109], treatment of RRMS patients with IL-12/23p40 neutralizing monoclonal Ab (ustekinumab) did not result in any significant therapeutic benefits in a phase II trial [110].
CD28/B7 costimulatory molecules as therapeutic targets
Optimal priming and activation of CD4+ T cells requires T cell receptor (TCR)-recognition of peptide-MHC class II complexes together with signals delivered by costimulatory molecules expressed on the surface of professional APCs. CD28 is constitutively expressed on 90% of naive and activated CD4+ T cells and on 50% of CD8+ T cells. By binding its cognate ligands B7.1/CD80 or B7.2/CD86 on the surface of professional APCs, CD28 lowers TCR activation threshold, thus leading to the augmentation of early signalling events necessary for efficient cytokine production, cell cycle progression and survival. Furthermore, CD28 ligation enhances the expression of CD40L and adhesion molecules necessary for trafficking, such as VLA-4. Moreover, CD28 is able to emanate TCR-independent autonomous signals, which account for its critical role in the regulation of proinflammatory cytokine/chemokine [111,112] and Th17 amplification in MS [113].
Another function of CD28 is its involvement in the development and homeostasis of Tregs. CD28 is required for both efficient generation of Tregs in the thymus [114] and for limiting T cell activation by sustaining the survival of Tregs in the periphery [115]. These evidences support the hypothesis that the presence of B7, even on cells that are not displaying the cognate antigen, may control adaptive immune responses [116]. Therefore, CD28 deficiency can lead to a reduced disease potential as well as to an enhanced susceptibility to autoimmune diseases by altering T cell effector and Tregs compartments, thus representing an interesting target to ameliorate cell dysfunctions in autoimmune diseases.
All CNS-infiltrating mononuclear cells in EAE, including CD4+ T cells, express CD80/B7.1 [117,118]. However, conflicting and opposing results have been obtained by using CTLA4-Ig or anti-B7 antibodies on EAE mice. Although, CTLA-Ig efficiently inhibits TCR-induced activation of autoreactive T cells by blocking CD28/ B7 interaction, it cross-links CD80 expressed on either APCs or activated autoreactive T cells, thus inducing IFN-γ production and contributing to the disease exacerbation [119]. Moreover, several studies evidenced that peripheral myelin-reactive T cells from MS patients are primarily effector/memory T cells in a higher activation state, which express high avidity TCRs and are less dependent from CD28/B7 costimulation in terms of TCR-activation [120].
Blockade of CD28/B7 interaction by recombinant CTLA4-Ig (adaptacept), which binds B7.1/B7.2 with high affinity, is being evaluated in RRMS in both phase I [121] and phase II clinical trials (ClinicalTrials.gov, identifier: NCT01116427).
More recently, anti-CD28 superagonistic Abs have been successfully used to inhibit the onset, progression and clinical course of EAE, by preferentially activating and expanding the immunosuppressive Tregs [122]. However, when administered to volunteers in phase I clinical trial, a humanized CD28 superagonistic Ab (TGN1412) induced a rapid and massive production of proinflammatory cytokines (i.e. IFN-γ and TNF-a), thus causing a severe systemic inflammatory response syndrome [123]. Moreover, our group has recently observed that the human superagonistic CD28 ANC28.1 Ab strongly up-regulated CD4+CD25highFOXP3+ Tregs at a similar extent in RRMS patients and healthy donors [113].
These data arise, at least, two important considerations: 1) CD28 signals can mediate different functions depending on the presence or not of TCR co-engagement and 2) the translation of experimental results from mice to men could determine dramatic effects, supporting differences in CD28 functions and signalling capability between humans and mice. Thus, the blockage of CD28 signalling, rather than its stimulation, may have heuristic implication for the development of more efficient therapies in MS.
CD28-associated PI3K signalling pathway as a potential therapeutic target
Phosphatidylinositol-3 kinase (PI3K) is a critical mediator of CD28 signalling [124]. The intracytoplasmic tails of CD28 binds and activates class 1 PI3K that phosphorylates inositol phospholipids on carbon atom 3, thus generating the phosphatidylinositol 3,4-biphosphate (PIP2) and phosphatidylinositol 3,4,5-triphosphate (PIP3) [112,125]. PIP2 and PIP3 lipids bind the pleckstrin domains of several molecules involved in CD28 signals, in particular the protein kinase B/Akt [126,127], thus affecting both the fate and movement through the body of T cells [128,129].
The PI3K/Akt pathway plays a central role in regulating inflammation and abnormalities in this pathway could be linked to the development of autoimmunity [130]. The activity of the PI3K network can be manipulated by pharmacological compounds, several of which are approved for clinical use in some inflammatory and autoimmune diseases. Blockage of class I PI3Kγ subunit has resulted effective inreducing both disease severity and incidence in a mouse model of systemic lupus erythematosus [131] and the delivery of oral active form of PI3Kγ inhibitor AS-605240 reduces joint inflammation and cartilage erosion in mouse models of rheumatoid arthritis [132,133].
Our recent results on the ability of AS-605240 to inhibit the proinflammatory responses induced by CD28 stimulation in RRMS, suggest PI3K as a valuable therapeutic candidate for MS. Moreover, the availability of oral active forms of PI3K inhibitors [134] may also circumvent the ASEs (e.g. skin indurations and lipoatrophy) related to infusion or injection and may exhibit higher adherence and long-term persistence, as observed for other oral compounds (e.g. fingolimod) [85].
Conclusion
Inflammatory T cells have a well recognized role in the pathogenesis of MS. Most of the current therapeutic strategies focus on blocking the pro-inflammatory responses in a non-specific manner, thus compromising the ability of T cells to fight infections or to control cancer. From basic researches and pre-clinical findings in animal model, a growing number of therapeutic agents are upcoming. A future challenge will be to identify the most efficacious, safe and tolerable anti- inflammatory drugs that used in combination with treatments that block neurodegeneration may also favour remyelination and tissue repair.
Acknowledgement
This literature review was fully supported by a grant from the “Fondazione Italiana Sclerosi Multipla” (Project No. FISM 2011/R/36).
References
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006; 354: 942-955.
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005; 23: 683-747.
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012; 8: 647-656.
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009; 132: 1175-1189.
- Batoulis H, Addicks K, Kuerten S. Emerging concepts in autoimmune encephalomyelitis beyond the CD4/T(H)1 paradigm. Ann Anat. 2010; 192: 179-193.
- Oksenberg JR, Hauser SL. Genetics of multiple sclerosis. Neurol Clin. 2005; 23: 61-75, vi.
- Hafler DA, Benjamin DS, Burks J, Weiner HL. Myelin basic protein and proteolipid protein reactivity of brain- and cerebrospinal fluid-derived T cell clones in multiple sclerosis and postinfectious encephalomyelitis. J Immunol. 1987; 139: 68-72.
- Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA, et al. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 1990; 346: 183-187.
- Zamvil S, Nelson P, Trotter J, Mitchell D, Knobler R, Fritz R, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985; 317: 355-358.
- Zamvil SS, Nelson PA, Mitchell DJ, Knobler RL, Fritz RB, Steinman L, et al. Encephalitogenic T cell clones specific for myelin basic protein. An unusual bias in antigen recognition. J Exp Med. 1985; 162: 2107-2124.
- Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2- responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994; 179: 973-984.
- Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, Lafaille JJ, et al. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol. 2008; 181: 4648-4655.
- Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R, et al. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004; 172: 3893-3904.
- Bielekova B, Lincoln A, McFarland H, Martin R. Therapeutic potential of phosphodiesterase-4 and -3 inhibitors in Th1-mediated autoimmune diseases. J Immunol. 2000; 164: 1117-1124.
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989; 7: 145-173.
- Romagnani S. The Th1/Th2 paradigm. Immunol Today. 1997; 18: 263-266.
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996; 156: 5-7.
- Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996; 26: 1641-1646.
- Segal BM, Shevach EM. IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med. 1996; 184: 771-775.
- Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000; 164: 2759-2768.
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002; 169: 7104-7110.
- Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003; 170: 2153-2160.
- Gran B, Chu N, Zhang GX, Yu S, Li Y, Chen XH. Early administration of IL-12 suppresses EAE through induction of interferon-gamma. J Neuroimmunol. 2004; 156: 123-131.
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000; 13: 715-725.
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003; 278: 1910-1914.
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005; 201: 233-240.
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17- producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005; 6: 1123-1132.
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005; 6: 1133-1141.
- Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010; 24: 1023-1034.
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007; 8: 942-949.
- Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007; 448: 484-487.
- Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008; 9: 641-649.
- Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008; 9: 650-657.
- Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008; 454: 350-352.
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007; 8: 967-974.
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008; 14: 337-342.
- Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009; 132: 3329-3341.
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008; 172: 146-155.
- Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH, et al. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008; 125: 161-169.
- Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011; 74: 1-13.
- Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005; 6: 331-337.
- Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006; 24: 209-226.
- Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006; 7: 401-410.
- Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol. 2008; 4: 384-398.
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007; 13: 423-431.
- Dalla Libera D, Di Mitri D, Bergami A, Centonze D, Gasperini C, Grasso MG, et al. T regulatory cells are markers of disease activity in multiple sclerosis patients. PLoS One. 2011; 6: e21386.
- Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. 2005; 81: 45-52.
- Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens JL, et al. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. 2008; 123: 79-89.
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004; 199: 971-979.
- Jacobs L, O'Malley J, Freeman A, Ekes R. Intrathecal interferon reduces exacerbations of multiple sclerosis. Science. 1981; 214: 1026-1028.
- Weinstock-Guttman B, Ransohoff RM, Kinkel RP, Rudick RA. The interferons: biological effects, mechanisms of action, and use in multiple sclerosis. Ann Neurol. 1995; 37: 7-15.
- Kappos L, Polman C, Pozzilli C, Thompson A, Beckmann K, Dahlke F; European Study Group in Interferon beta-1b in Secondary-Progressive MS. Final analysis of the European multicenter trial on IFNbeta-1b in secondary-progressive MS. Neurology. 2001; 57: 1969-1975.
- Rojas JI, Romano M, Ciapponi A, Patrucco L, Cristiano E. Interferon beta for primary progressive multiple sclerosis. Cochrane Database Syst Rev. 2009; CD006643.
- Jiang H, Milo R, Swoveland P, Johnson KP, Panitch H, Dhib-Jalbut S, et al. Interferon beta-1b reduces interferon gamma-induced antigen-presenting capacity of human glial and B cells. J Neuroimmunol. 1995; 61: 17-25.
- Genç K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. 1997; 99: 2664-2671.
- Teleshova N, Bao W, Kivisäkk P, Ozenci V, Mustafa M, Link H, et al. Elevated CD40 ligand expressing blood T-cell levels in multiple sclerosis are reversed by interferon-beta treatment. Scand J Immunol. 2000; 51: 312-320.
- Krakauer M, Sorensen P, Khademi M, Olsson T, Sellebjerg F. Increased IL-10 mRNA and IL-23 mRNA expression in multiple sclerosis: interferon-beta treatment increases IL-10 mRNA expression while reducing IL-23 mRNA expression. Mult Scler. 2008; 14: 622-630.
- Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009; 183: 5418-5427.
- Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC, et al. IL-27 mediates the response to IFN-β therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun. 2011; 25: 1170-1181.
- Chen M, Chen G, Deng S, Liu X, Hutton GJ, Hong J, et al. IFN-β induces the proliferation of CD4+CD25+Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J Neuroimmunol. 2012; 242: 39-46.
- Namdar A, Nikbin B, Ghabaee M, Bayati A, Izad M. Effect of IFN-beta therapy on the frequency and function of CD4(+)CD25(+) regulatory T cells and Foxp3 gene expression in relapsing-remitting multiple sclerosis (RRMS): a preliminary study. J Neuroimmunol. 2010; 218: 120-124.
- Río J, Nos C, Tintoré M, Téllez N, Galán I, Pelayo R. Defining the response to interferon-beta in relapsing-remitting multiple sclerosis patients. Ann Neurol. 2006; 59: 344-352.
- Bertolotto A. Neutralizing antibodies to interferon beta: implications for the management of multiple sclerosis. Curr Opin Neurol. 2004; 17: 241-246.
- Valenzuela RM, Costello K, Chen M, Said A, Johnson KP, Dhib-Jalbut S, et al. Clinical response to glatiramer acetate correlates with modulation of IFN-gamma and IL-4 expression in multiple sclerosis. Mult Scler. 2007; 13: 754-762.
- Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. 2003; 170: 4483-4488.
- Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B, et al. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J Neuroimmunol. 2009; 216: 113-117.
- Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci U S A. 2005; 102: 6449-6454.
- Mikol DD, Barkhof F, Chang P, Coyle PK, Jeffery DR, Schwid SR, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008; 7: 903-914.
- O'Connor P, Filippi M, Arnason B, Comi G, Cook S, Goodin D, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009; 8: 889-897.
- Ghalie RG, Edan G, Laurent M, Mauch E, Eisenman S, Hartung HP, et al. Cardiac adverse effects associated with mitoxantrone (Novantrone) therapy in patients with MS. Neurology. 2002; 59: 909-913.
- Ghalie RG, Mauch E, Edan G, Hartung HP, Gonsette RE, Eisenmann S, et al. A study of therapy-related acute leukaemia after mitoxantrone therapy for multiple sclerosis. Mult Scler. 2002; 8: 441-445.
- Hartung H1, Gonsette R, König N, Kwiecinski H, Guseo A, Morrissey SP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002; 360: 2018-2025.
- Gasperini C, Ruggieri S. New oral drugs for multiple sclerosis. Neurol Sci. 2009; 30 Suppl 2: S179-183.
- Gasperini C, Ruggieri S. Emerging oral drugs for relapsing-remitting multiple sclerosis. Expert Opin Emerg Drugs. 2011; 16: 697-712.
- Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992; 356: 63-66.
- Stuve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol. 2006; 63: 1383-1387.
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006; 354: 899-910.
- Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006; 354: 911-923.
- Phillips JT, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. Infusion-related hypersensitivity reactions during natalizumab treatment. Neurology. 2006; 67: 1717-1718.
- Baldwin KJ, Hogg JP. Progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Curr Opin Neurol. 2013; 26: 318-323.
- Jackson SJ, Giovannoni G, Baker D. Fingolimod modulates microglial activation to augment markers of remyelination. J Neuroinflammation. 2011; 8: 76.
- Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP, et al. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008; 63: 61-71.
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010; 362: 402-415.
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010; 362: 387-401.
- Gasperini C, Ruggieri S. Development of oral agent in the treatment of multiple sclerosis: how the first available oral therapy, fingolimod will change therapeutic paradigm approach. Drug Des Devel Ther. 2012; 6: 175-186.
- Espinosa PS, Berger JR. Delayed fingolimod-associated asystole. Mult Scler. 2011; 17: 1387-1389.
- Lindsey JW, Haden-Pinneri K, Memon NB, Buja LM. Sudden unexpected death on fingolimod. Mult Scler. 2012; 18: 1507-1508.
- Carruthers RL, Rotstein DL, Healy BC, Chitnis T, Weiner HL, Buckle GJ, et al. An observational comparison of natalizumab vs. fingolimod using JCV serology to determine therapy. Mult Scler. 2014; 20: 1381-1390.
- Beutler E, Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, et al. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci U S A. 1996; 93: 1716-1720.
- Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, Beutler E. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet. 1994; 344: 9-13.
- Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, Beutler E. Development of cladribine treatment in multiple sclerosis. Mult Scler. 1996; 1: 343-347.
- Cook S, Vermersch P, Comi G, Giovannoni G, Rammohan K, Rieckmann P, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler. 2011; 17: 578-593.
- Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010; 362: 416-426.
- Giovannoni G, Cook S, Rammohan K, Rieckmann P, Sorensen PS, Vermersch P, et al. Sustained disease-activity-free status in patients with relapsing-remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis. Lancet Neurol. 2011; 10: 329-337.
- Yates D. Orally administered cladribine displays efficacy in multiplesclerosis trial. Nat Rev Neurol. 2010; 6: 182.
- Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha- targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006; 103: 5941-5946.
- Hao J, Campagnolo D, Liu R, Piao W, Shi S, Hu B, et al. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol. 2011; 69: 721-734.
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013; 381: 2167-2175.
- Oh J, Saidha S, Cortese I, Ohayon J, Bielekova B, Calabresi PA, et al. Daclizumab-induced adverse events in multiple organ systems in multiple sclerosis. Neurology. 2014; 82: 984-988.
- Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010; 9: 381-390.
- Gilleece MH, Dexter TM. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood. 1993; 82: 807-812.
- Coles AJ1, Cox A, Le Page E, Jones J, Trip SA, Deans J. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006; 253: 98-108.
- Riechmann L1, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988; 332: 323-327.
- Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012; 380: 1819-1828.
- Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012; 380: 1829-1839.
- Daniels GH, Vladic A, Brinar V, Zavalishin I, Valente W, Oyuela P, et al. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab. 2014; 99: 80-89.
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006; 116: 1317-1326.
- Ichikawa M, Koh CS, Inoue A, Tsuyusaki J, Yamazaki M, Inaba Y, et al. Anti-IL-12 antibody prevents the development and progression of multiple sclerosis-like relapsing-- remitting demyelinating disease in NOD mice induced with myelin oligodendrocyte glycoprotein peptide. J Neuroimmunol. 2000; 102: 56-66.
- Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med. 1995; 181: 381-386.
- Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008; 7: 796-804.
- Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-kappaB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci U S A. 2004; 101: 6098-6103.
- Tuosto L. NF-κB family of transcription factors: biochemical players of CD28 co-stimulation. Immunol Lett. 2011; 135: 1-9.
- Camperio C, Muscolini M, Volpe E, Di Mitri D, Mechelli R, Buscarinu MC, et al. CD28 ligation in the absence of TCR stimulation up-regulates IL-17A and pro-inflammatory cytokines in relapsing-remitting multiple sclerosis T lymphocytes. Immunol Lett. 2014; 158: 134-142.
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000; 12: 431-440.
- Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003; 171: 3348-3352.
- Lohr J, Knoechel B, Jiang S, Sharpe AH, Abbas AK. The inhibitory function of B7 costimulators in T cell responses to foreign and self-antigens. Nat Immunol. 2003; 4: 664-669.
- Racke MK, Scott DE, Quigley L, Gray GS, Abe R, June CH, et al. Distinct roles for B7-1 (CD-80) and B7-2 (CD-86) in the initiation of experimental allergic encephalomyelitis. J Clin Invest. 1995; 96: 2195-2203.
- Windhagen A, Newcombe J, Dangond F, Strand C, Woodroofe MN, Cuzner ML, et al. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995; 182: 1985-1996.
- Perrin PJ, Scott D, Davis TA, Gray GS, Doggett MJ, Abe R. Opposing effects of CTLA4-Ig and anti-CD80 (B7-1) plus anti-CD86 (B7-2) on experimental allergic encephalomyelitis. J Neuroimmunol. 1996; 65: 31-39.
- Bar-Or A. The immunology of multiple sclerosis. Semin Neurol. 2008; 28: 29-45.
- Viglietta V, Bourcier K, Buckle GJ, Healy B, Weiner HL, Hafler DA, et al. CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology. 2008; 71: 917-924.
- Lin CH, Hünig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol. 2003; 33: 626-638.
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006; 355: 1018-1028.
- Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009; 229: 12-26.
- Muscolini M, Camperio C, Capuano C, Caristi S, Piccolella E, Galandrini R, et al. Phosphatidylinositol 4-phosphate 5-kinase α activation critically contributes to CD28-dependent signaling responses. J Immunol. 2013; 190: 5279-5286.
- Garçon F, Patton DT, Emery JL, Hirsch E, Rottapel R, Sasaki T. CD28 provides T-cell costimulation and enhances PI3K activity at the immune synapse independently of its capacity to interact with the p85/p110 heterodimer. Blood. 2008; 111: 1464-1471.
- Okkenhaug K, Patton DT, Bilancio A, Garçon F, Rowan WC, Vanhaesebroeck B, et al. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol. 2006; 177: 5122-5128.
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008; 105: 7797-7802.
- Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008; 9: 513-521.
- Ghigo A, Damilano F, Braccini L, Hirsch E. PI3K inhibition in inflammation: Toward tailored therapies for specific diseases. Bioessays. 2010; 32: 185-196.
- Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005; 11: 933-935.
- Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005; 11: 936-943.
- Hayer S, Pundt N, Peters MA, Wunrau C, Kühnel I, Neugebauer K, et al. PI3Kgamma regulates cartilage damage in chronic inflammatory arthritis. FASEB J. 2009; 23: 4288-4298.
- Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012; 11: 317-328.