
Research Article
Austin Neurosurg Open Access. 2014;1(3): 1015.
Surface Protein Dynamics in Glioma Stem Cells
Soeda A1*, Ohe N1, Lee D2, Iwama T1 and Park DM2
1Department of Neurosurgery, Gifu University School of Medicine, Japan
2Department of Neurological Surgery, University of Virginia, USA
*Corresponding author: Soeda A, Department of Neurosurgery, Gifu University School of Medicine, 1-1 Yanagido, Gifu City, Gifu 501-1194, Japan
Received: May 14, 2014; Accepted: June 05, 2014; Published: June 05, 2014
Abstract
Introduction: Flow cytometry is used for isolating Cancer Stem Cells (CSCs) by recognizing their surface markers such as CD133, CD44, CD24, and CD15. Although several CSC markers are helpful to identify CSCs, it remains unknown whether these markers can be used only when freshly derived from surgical tissue specimens or even after long–term exposure to the in vitro environment.
Material and Methods: We established and evaluated 8 glioma cell lines from glioblastoma multiforme tissue specimens. Flow cytometry was used to analyze the stem cells, glia, and neuronal– and cell–adhesion molecules at the following time periods: (1) within 3 months of tumorsphere culturing (primary analysis), (2) between the stage of dissociated single cells and subsequently developed spheres at the same passage period (passage analysis), and (3) 3–36 months after primary analysis (mid⁄long–term analysis). We also evaluated the differences in the surface markers after differentiation following serum addition.
Results: The primary analysis of the fresh surgical tissue specimens revealed different expression patterns, especially for CD24 (5–99%) and CD184 (3–40%), with higher expression levels for CD44 and CD146. After sphere development from the dissociated single cells, the CD54 expression was elevated with epidermal growth factor receptor (EGFR) degradation during cell passages. After serum addition, the CD133, A2B5, CD24, CD56, and CD184 expression decreased, suggesting the potential of these proteins as stemness markers. At long–term analysis, most surface markers were found to be stable; however, the expression profiles of several markers differed among the CSC lines.
Conclusion: Glioma CSCs maintain stable expression of stem cell markers even under long–term in vitro propagation. Our results may facilitate identification of novel cell markers for application in the diagnosis and treatment of gliomas.
Keywords: Glioma; Glioma stem cell; Flow cytometry; CD133; Cell adhesion molecule
Introduction
Cancer Stem Cells (CSCs) have been identified in several malignancies, and a variety of experimental approaches have been initiated to analyze their properties [1]. Comprehending the unique properties of CSCs is a high priority for researches aimed at elucidating the molecular mechanisms driving tumor initiation and for the development of therapeutic strategies specifically targeting CSC population [2]. Recent advances in stem cell biology, cell signaling, computational technology, and genetic model systems have revolutionized our understanding of the mechanisms underlying the genetics, biology, and clinical behavior of cancer [3]. Among these, flow cytometry technique can be applied for the isolation of CSCs by recognizing the CSC surface markers such as CD133, CD44, CD24, and CD15 [4–6].
CD133 is the most commonly used glioma CSC marker for studying aspects such as in vivo–tumor formation ability [4]. Although the function of CD133 remains unknown, it has proven useful in several other solid cancers such as colorectal cancers [7]. However, several reports have suggested a less clear distinction between the abilities of CD133+ and CD133− cells to form tumors [8,9]. Several strategies have been used for the identification of CSCs of gliomas and other cancers such as colon, prostate, and lung cancers. For example, CD44 has been identified as a potential breast and prostate CSC marker [5,10]. Moreover, the expression profile of CD44 also identifies the astrocytic progenitors, and most gliomas express this marker [11,12]. High CD24 expression level helps identify transitamplifying cells as well as differentiated neurons, and CD24 is also required for the terminal differentiation of neuronal progenitors [13]. Furthermore, A2B5–expressing glial–restricted precursor is capable of generating oligodendrocytes astrocytes and gliomas [9]. CD184 is a chemokine receptor involved in Neural Stem Cell (NSC) migration; it has been implicated in the invasion of gliomas and metastasis of pancreatic CSCs [14]. Moreover, several CAMs are also involved in gliomagenesis [15,16]. However, it remains unknown whether thesemarkers are useful only when used immediately after derivation from surgical specimens or even after long–term exposure to the in vitro environment.
Patient–specific CSC lines are a powerful tool in the study of CSC biology that can be exploited for the development of therapies targeted at specific patients. However, CSCs may adapt differently to the prevailing culture conditions or may reflect the intrinsic genetic fluctuations typical of tumor cells [17–19]. The isolation and manipulation of CSCs may introduce artifacts under in vitro conditions at primary analysis, long–term analysis, and under in vivo microenvironment [20]. Alternatively, CSCs may be reprogrammed in long–term in vitro culture settings or may dedifferentiate in vitro from a more differentiated cell type in response to certain signal transduction cascades [21]. Therefore, to address these issues, it is important to characterize CSC lines from different patients at different time periods.
In this study, we evaluated several surface markers obtained from surgical tissue specimens and glioma CSCs derived from patients by the neurosphere assay [19]. We compared the surface–marker dynamics at the following time periods: (1) within 3 months of tumorsphere culturing (primary analysis), (2) between the stages of dissociated single cells and subsequently developed spheres (during the same passage), and (3) 6–36 months after primary analysis (middle to long–term analysis). To minimize the influence of artificial in vitro effects, long–term analysis was performed at least twice and their results were averaged. Because CSCs can differentiate in the presence of ideal compounds such as retinoic acid, bone morphogenetic protein, and serum, a better understanding of the makers expressed in differentiated CSCs will be useful in CSC biology [18,22]. With this perspective, we evaluated the surface–marker differences after differentiation following serum addition.
Material and Methods
Cell culture
Tumorsphere culturing was performed as described previously with some modifications in the medium. We used Dulbecco’s modified Eagle’s medium⁄nutrient mixture F–12 (DMEM–F12; GIBCO–Invitrogen, La Jolla, CA) supplemented with penicillin G, streptomycin sulfate, B–27 (GIBCO–Invitrogen), recombinant human FGF–2 (20 ng⁄mL; R&D Systems, Minneapolis, MN), and recombinant human epidermal growth factor (EGF; 20 ng⁄mL; R&D Systems) [19]. The cells were cultured in HERA cell incubators (Thermo Electronic Corporation, Asheville, NC) at 37°C, ≥95% relative humidity, and 5% CO2 with 20% O2 conditions. Prior informed consent was obtained from the donor patients. Our study was approved by the Medical Review Boards of University of Pittsburgh, University of Virginia, and Gifu University School of Medicine.
Flow cytometry
For flow cytometry of the surgical specimens within 2 h of tumor removal, the tumor tissues were minced by a surgical scalpel, incubated in Accutase (Sigma–Aldrich, St. Louis, MO) for 20 min under 37°C and washed; the cells were then dissociated in phosphatebuffered saline (PBS; 3X) to remove the cell debris, and titrated in PBS. The cells were then passed through a 40–μm strainer (Falcon, Oxnard, CA) and resuspended in flow cytometry buffer consisting of PBS with 0.1% fraction V of bovine serum albumin (Sigma–Aldrich). The sphere cells were mechanically dissociated by a 5–mL pipette (Corning, NY) and then passed through a 40–μm strainer (Falcon). For long–term analysis, we used clonally expanded frozen–cultured X01, X02, and X03 sphere cells at different time points [18,19]. Briefly, the cells were diluted to 1 × 105 concentration with 50–μL aliquots for each analysis. For surface–marker analysis, we used antibodies against anti–human phycoerythrin (PE)–conjugated anti–human CD184, CD44, CD24, CD15, PDGFRa, CD54 (or intracellular adhesion molecule–1, ICAM–1), CD56 (or neural CAM, NCAM), CD146 (or melanoma CAM, MCAM), CD166 (or activated–leukocyte CAM, ALCAM), EGFR (BD Biosciences, San Jose, CA), PE–conjugated CD133⁄1 (AC133) (Miltenyi Biotec, Auburn, CA), and purified anti–human A2B5 (Miltenyi Biotec). Antibodies were titrated using appropriate dilutions and incubated on an ice bath for 60 min. The cells were then washed with the flow cytometry buffer, and the secondary fluorescent–conjugated antibody for A2B5 was added at appropriate dilutions and incubated on an ice bath for 60 min. For intracellular staining, the cell pellets were incubated with 0.1% Triton X–100 in the flow cytometry buffer on an ice bath for 10 min and then washed with the flow cytometry buffer. Sox2 (R&D Systems) and bmi– 1 (R&D Systems) were used for intracellular staining. The stained cells were washed once with the flow cytometry buffer, resuspended in 500 μL of the same buffer, and evaluated by the Coulter EPICS Cytometer (Beckman Coulter, Fullerton, CA). Appropriate compensation and isotype controls were used in the experiment.
Immunofluorescent staining
Immunocytochemistry of CSCs was performed as described in a previous study [23]. The following antibodies were used: antinestin (rabbit pAb, 1:200; Chemicon, Temecula, CA), anti–CD133⁄1 (1:1; Miltenyi Biotec), anti–ß–III–tubulin (Tuj1; mouse mAb, 1:200; Chemicon) for neurons, and anti–glial fibrillary acidic protein (GFAP; rabbit pAb, 1:500; DAKO, Glostrup, Denmark) for astrocytes. Visualizations were performed with Alexa fluorophore–conjugated secondary antibodies (1:1,000; Molecular Probes, Eugene, OR).
Statistical analysis
The differences among the various surface maker expression patterns were evaluated by Student’s t–test. p < 0.05 was considered statistically significant.
Results
Glioma CSCs and glial⁄neural linage markers of freshly dissected brain tumors and primary–established glioma CSCs
All experiments were performed with malignant–glioma derived CSC lines from freshly resected surgical specimens [19,23]. Our culture system allowed the isolation of clonogenic cells from the human brain tumors and that these tumors contained multipotent, long–term self–renewing, population–expanding cells that satisfy the defining criteria of CSCs [1,4,18]. We analyzed the expression patterns of surface markers CD44, A2B5, and PDGFR–a for glia, CD24 for neural cells, and CD133 and CD15 for stem cells on fresh glioblastoma multiforme (GBM) specimens by flow cytometry. The CD184 expression was also analyzed because it is extremely important for tumor invasion. High CD44 expression was observed in all 8 cases and high CD24 expression was observed in 3 cases (1203, 0320, and 0408) (Table 1). The cells from all 8 GBM samples formed neuronal sphere–like aggregates within 2 to 7 days of culturing. The spherelike aggregation of cells from cell lines 1203 (X04), 0320 (X06), and 0408 (X07) increased continuously, while the cells from the other 5 samples became adherent and lost their proliferative capacity within 3 months of culturing. X04, X06, and X07 could be passaged and amplified by resupplementing with fresh medium twice weekly. Interestingly, in these 3 cases, the CD24 expression was nearly 100%, while the CD184 expression was higher than that in other cases (p < 0.01). No correlation was noted between the CD133 expression and cell amplification.
![]()
Sample-ID
Alternative ID
Diagnosis
WHO grade
Marker Expression (%)
CXCR4
CD133
CD44
CD24
A2B5
PDGFRa
CD15
0917
GBM
IV
1.0
4.8
98.4
11.8
12.7
21.9
2.2
0815
GBM
IV
1.4
1.1
90.4
1.1
8.6
0.6
1.8
0626
GBM
IV
2.0
1.7
67.4
0.8
7.1
1.3
0.9
1203
X04
GBM
IV
16.4
1.7
98.5
98.4
1.1
0.5
1.4
0320
X06
GBM
IV
23.5
3.2
97.6
96.5
1.5
0.8
0.2
0408
X07
Gliosarcoma
IV
15.6
5.6
98.9
92.5
0.1
1.1
0.2
0609
GBM
IV
2.2
1.9
76.5
16.2
8.2
19.1
2.3
0630
GBM
IV
7.9
2.5
89.8
25.2
6.2
4.2
0.5
Table 1: The values indicate the percentage expression of surface markers. Abbreviation: GBM, Glioblastoma multiforme.
After sufficient amplification of these 3 cases, we analyzed the expression patterns of several surface markers. We also analyzedthe established glioma CSC lines X01, X02, and X03 and the newly established GBM sphere cells X08 and X09. Moreover, CD56 as a neuronal marker and CD54, CD146, and CD166 as cell–surface markers are expressed in glioma⁄cancers. In addition, we found that the EGFR expression was expressed in immature astrocytes and was essential for astrocyte development. EGFR was also frequently over expressed in high–grade glioma cells. The stem cell markers Sox2 and bmi–1 that are highly expressed in glioma CSCs were analyzed by intra–cellular staining method. All cases revealed high expression levels of Sox2, EGFR, bmi–1, and CD146, while the expression profiles of CD133, CD44, CD24, and CD56 differed among tumors (Table 2). Although CD15 has been reported to be a glioma CSC marker, we detected only few CD15 cells in this study [6].
![]()
Case ID
TCGA Data
Marker expression (%)
X01
X02
X03
X04
X06
X07
X08
X09
% Expressions of cases
Glioma CSC Marker
CD133
4.9
2.6
9.3
2.8
3.6
4.6
1
1.5
81
CD15 (SSEA1)
0.4
1
0.4
0.7
1.5
1.3
0.1
0.5
2
Sox2 (ICS)
97.4
92.1
91.3
97.2
95.6
97
98
92.1
96
Bmi 1 (ICS)
64.6
52.1
42.5
53.2
44.2
40.1
56.7
36.2
30
Glial Marker
CD44 (HCAM)
88.2
9.7
94.7
94.5
99
98.3
92.5
81.4
98
A2B5
6.5
9.2
12.1
4.3
9.3
1.2
11
5.5
N/A
PDGFRa (CD140a)
0.8
0.5
0.7
0.3
1.4
0.8
1.1
0.7
15
Neuronal Marker
CD24
7.4
99.8
46.5
97.1
99.2
97.2
85.3
7.7
N/A
NCAM (CD56)������������ 63.9���� 31.7����������� 80
89.4
95.4
57.4
43
57
38.4
63.9
31.7
80
Others
CXCR4 (CD186)
40.9
15.1
51.2
10.1
10.5
9.8
7.3
7.5
97
ICAM-1 (CD54)
97.8
89.7
91.1
39
65
47
38.7
23.6
50
MCAM (CD 146)
96.2
89.2
76.5
98
92.1
88.7
61.7
64.9
89
ALCAM (CD 166)
23.5
69.7
59.8
41.2
35.8
68.6
67.2
61.8
63
EGFR
53
64.2
39.1
47.7
52
51.7
36
37.3
75
Table 2: The values indicate the percentage expression of surface markers. Flow cytometry data were collected within 3 months of amplifying the cells. The TCGA data is given in the left panel. The numerals indicate percent positive cases from the database.
Marker changes between dissociated single cells and subsequently developed spheres and differentiated glioma CSCs
The in vitro environments are different inside and outside of the sphere cells during development [24]. Therefore, we first evaluated the marker expression patterns of dissociated single sphere cells prior to the addition of EGF and 72 h later for the developed sphere cells. The dissociated sphere cells, mostly in single–cell suspension, formed spheres within 24 h, and their size increased (Figure 1A). In addition, EGF concentration was decreased (data not shown). As expected, the EGFR expression decreased after 72 h of culturing (Figure 1B). Although most of the surface markers were expressed in similar levels, the level of CD54 expression was increased, but it returned to the original after addition of EGF and after dissociation during the subsequent passage in all cell lines.
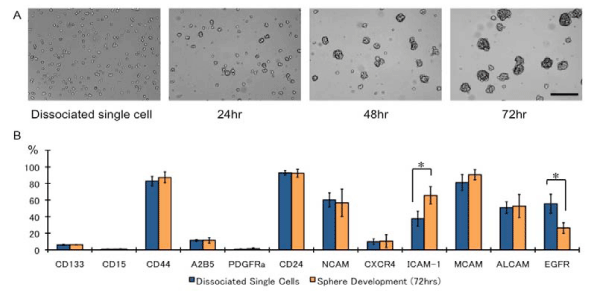
Figure 1: The time course of developing spheres and surface-marker differences. (A) Dissociated single cells formed spheres within 24 h. Bar: 100 μm, (B) Surface maker differences between dissociated single cells and subsequently developed spheres after 72 h of culturing. The sphere cells were mechanically dissociated and analyzed. N = 3, *p < 0.01.
In the serum–containing differentiation medium, the sphere cells differentiated and developed into neurons and glia, expressing both neuronal and glia markers (Figure 2A). Analysis of the markers n the differentiated cells revealed reduced CD133 expression after differentiation; this observation is in concordance with that of a previous study [18]. The A2B5, CD24, CD56, and CD184 expression also dramatically decreased after differentiation, and the CD166 expression increased (p < 0.05). These data suggest that A2B5, CD24, CD56, and CD184 may be glioma stem⁄progenitor cells and that CD166 may be a differentiation marker.
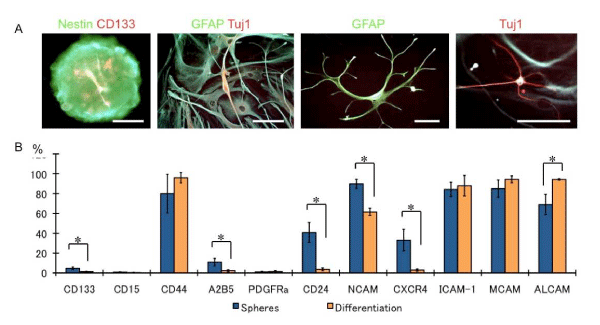
Figure 2: Glioma CSCs differentiation following serum addition. (A) Nestin and CD133-positive sphere cells (left panel) differentiated GFAP-positive glia and Tuj1-positive neurons. Some cells showed GFAP and Tuj1 doublepositive cells. Bars: 25 μm,(B) Bar graph shows the marker expression patterns of the undifferentiated sphere cells and differentiated cells. N = 3, *p < 0.01.
Marker expression patterns of long–term–cultured glioma CSCs
To avoid any potential surface–marker differences between single cells and developed spheres, long–term flow cytometry was performed after dissociating the spheres and prior to adding EGF. Repeated characterization of the cell–surface phenotypes of sphere cells from X04, X06, and X07 by flow cytometry revealed that the cells remained stable up to 6 months (Table 3), while the bmi–1 expression was found to be increased in all 3 cell lines. Similarly, repeated evaluation of X01, X02, and X03 cells revealed that they remained stable for up to 36 months (Table 4). Surprisingly, in X01 cells, the CD24 expression dramatically increased after 12 months and further increased after 24 months of culturing. In X02 cells, the CD44 expression gradually increased in 6–24 months. However, the CD24 expression was decreased after 6 months. Interestingly, the self–renewal ability of X02 cells after 6 months of culturing was 3–times greater than that of the early passage cells (data not shown). On the other hand, nodifferences were observed in the X01 cells in terms of morphology, self–renewal ability, and tumorigenicity after intracranial injection [18].
![]()
X04
X06
X07
Marker expression (%)
3months
6months
3months
6months
3months
6months
Glioma CSC Marker
CD 133
2.4
2.8
1.1
1.8
1.8
2.8
CD15 (SSEA1)
0.2
0.2
1.2
1.1
0.7
1.1
Sox2 (ICS)
95.6
97.6
96.0
91.3
97.5
98.0
Bmil (ICS)
55.7
81.8
54.2
86.0
36.1
44.5
Glial Marker
CD44 (HCAM)
97.3
86.8
98.8
88.9
92.7
86.4
A2B5
4.9
7.7
6.4
12.2
1.9
1.8
PDGFRa (CD140a)
0.4
0.6
1.1
0.6
0.9
0.9
Neuronal Marker
CD24
93.5
93.5
97.0
97.4
91.0
85.3
NCAM (CD56)
48.6
44.0
61.9
64.7
34.8
50.0
Others
CXCR4 (CD186)
12.7
6.3
11.6
13.7
10.2
10.6
ICAM-1 (CD54)
40.0
32.2
53.7
46.1
24.3
23.5
MCAM (CD146)
97.5
88.7
93.9
84.9
79.4
56.6
ALCAM (CD166)
54.2
75.4
40.2
52.7
61.3
38.6
EGFR
30.9
43.2
66.4
59.7
55.6
67.8
Table 3: Marker Expressions in Middle-term Cultured Glioma CSCs.
![]()
X01
X02
X03
Marker expression (%)
3-6
months6-12
months12-36
months3-6
months6-12
months12-36
months3-6
months6-12
months12-36
monthsGlioma CSC Marker
CD 133
5.0
4.1
6.4
2.3
6.7
2.6
7.4
5.3
2.9
CD15 (SSEA1)
0.2
1.1
ND
1.4
0.8
ND
0.3
0.3
ND
Sox2 (ICS)
85.3
ND
93.2
94.0
ND
96.3
86.0
ND
91.2
Bmil (ICS)
78.4
ND
78.6
69.1
ND
72.3
59.2
ND
65.4
Glial Marker
CD44 (HCAM)
90.4
87.4
100.0
11.5
42.1
93.5
96.0
91.9
99.9
A2B5
8.7
3.0
11.8
23.3
11.2
10.8
18.8
5.9
9.5
PDGFRa (CD140a)
0.2
ND
ND
0.2
ND
ND
1.5
ND
ND
Neuronal Marker
CD24
10.4
7.1
61.9
99.3
73.8
11.0
37.6
43.9
43.9
NCAM (CD56)
92.4
99.8
73.4
92.1
96.0
90.2
65.9
54.2
61.0
Others
CXCR4 (CD186)
44.0
29.3
41.4
16.7
30.5
20.3
50.4
32.7
41.9
ICAM-1 (CD54)
96.8
95.6
95.0
94.9
89.4
81.1
96.6
94.5
73.3
MCAM (CD146)
90.8
91.3
89.8
83.6
92.1
92.6
89.5
94.2
41.2
ALCAM (CD166)
16.4
14.7
30.4
71.4
88.9
68.4
68.7
77.9
38.6
EGFR
47.1
44.0
42.0
57.0
58.0
42.0
61.0
44.0
46.0
Table 4: Marker Expressions in Long-term Cultured Glioma CSCs.
Discussion
Although the molecular biology aspect of cancer has been extensively studied, there is a lack of understanding of its cellular biology; for instance, the recently proposed cancer hierarchy and heterogeneity theories based on stem cell biology [1]. Stem cells occupy the top of the developmental hierarchy because they possess the ability of self–renewal and initiate development of all cell lineages in the corresponding tissues [25]. Stem cells divide to produce 2 daughter cells, of which one remains a stem cell and hence retains the self–renewal ability. The other daughter cell forms a progenitor cell that undergoes expansion and further differentiation to form mature cells. Because cancer develops as a result of unregulated self–renewal and differentiation, understanding cancer heterogeneity is fundamental to the understanding of cancer cell proliferation [26,27]. In fact, GBM displays a rather heterogeneous cellular composition, as indicated by the term “multiforme”; it exhibits phenotypic heterogeneity as it iscomposed of cells expressing both undifferentiated and differentiated markers [28]. In this study, clonally derived CSC lines from an individual patient expressed stem⁄progenitor markers such as CD133, CD44, CD24, and A2B5, which suggests that each tumor contained a fundamental type of stem and⁄or progenitor cell. However, the degree of differentiation and the types of cells produced were different for different tumors, which suggests a fundamental difference among the progenitor cells of different tumors [26,27].
Glioma CSCs express unique sets of molecular markers. Several stem⁄progenitor markers such as CD133, CD44, CD24, CD15, A2B5, and PDGFRa have been utilized for the identification and characterization of both normal stem cells and CSCs [4,6,11,12]. CD44 is one of the well–studied CAMs in cancer research, with a possible role in gliomagenesis [11,12,29]. CD171 (or neural CAM, L1CAM) is required for the maintenance of glioma CSCs [15]. However, very little is known about the possible role of other CAMs in gliomagenesis. In this study, we hypothesized that some CAMs expressing cell surface markers may be involved in the characterization of glioma heterogeneity. Thus, we selected CD54, CD56, CD146, and CD166. In fact, with reference to The Cancer Genome Atlas database (TCGA, http:⁄⁄cancergenome.nih.gov⁄), the expression of CD54 was detected in 50%, CD56 in 80%, CD146 in 89%, and CD166 in 63% of the GBM patients (Table 2). Furthermore, CD54 is associated with glioma invasion and several cancer prognoses [16]. In this study, the expression of CD54 ranged from 23.6–97.8% in the primary analysis, and the expression profile remained stable in long–term culturing. Interestingly, the CD54 expression increased with developing spheres, indicating the contribution of heterogeneous sphere environment in CD54 expression [24]. Neuronal lineage–restricted precursors can be differentiated and isolated via CD56 expression; this molecule is a type of neuronal marker such as CD24 [30]. Although no correlation was found between CD24 and CD56 expression in the sphere cultures, the CD24 expression decreased dramatically, while the CD56 expression was intact after differentiation following serum addition. These data suggest that CD24 and CD56 markers play different roles in neurogenesis and tumorigenesis. CD166 is also associated with malignancy in breast cancer and melanoma [31]; its expression differed and increased after differentiation, suggesting that CD166 may serve as a differentiation marker of stem⁄progenitor cells.
This study had certain limitations. In the primary cell culture, the cells from 5 of the 8 human tumor samples generated poorly expanding spheres, because of which we could not serially passage the cells growing in the suspension sphere culture. It is also possible those CSCs are reprogrammed during culture or that they dedifferentiate in vitro from a more differentiated cell type in response to certain signal–transduction cascade(s) [17,19,21]. Presently, we cannot rule out the possibility that transformation and dedifferentiation of more mature brain cells may contribute to tumorigenesis. It is intriguing to speculate whether specific growth factors can force lineagerestricted tumor stem cells to differentiate via different pathways. It is important to study this aspect further, because understanding the molecular basis of unregulated self–renewal of cancer cells will allow designing of more effective therapies. Moreover, it is important to study human specimens for studying CSCs, because CSCs derived from individual patients may facilitate better understanding of the origin⁄proliferation of various cancers.
In summary, we demonstrated that the established sphere culture method can preserve stem⁄progenitor marker expression even after 3 years of culturing. Our results also demonstrate the heterogeneity of glioma cell lines that may be involved in the differentiation⁄ dedifferentiation of CSCs in vitro, there by laying the foundation for further CSC studies aimed at clarification of their roles in the development of malignant brain tumors. A detailed understanding of the heterogeneity of CSCs will enable better understanding of the tumor pathogenesis and better designing of effective therapeutic strategies. In addition, to resolve the different surface–marker expression patterns among cancer patients, flow cytometry can be used as it allows characterization of more than 20 fluorescencelabeling. Thus, specific combination of cell surface molecules may enable characterization of CSC heterogeneity or CSC hierarchy in the future.
Acknowledgment
This work was supported by grants from JSPS KAKENHI (Grant Number 26670640) and the Department of Neurosurgery, Gifu University School of Medicine to A.S. and N.O.
References
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414: 105-111.
- Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007; 67: 8980-8984.
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9: 157-173.
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004; 432: 396-401.
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100: 3983-3988.
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009; 4: 440-452.
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007; 445: 106-110.
- Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, et al. Oct-4 Expression Maintained Cancer Stem-Like Properties in Lung Cancer-Derived CD133-Positive Cells. Plos One. 2008; 3: e2637.
- Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery. 2008; 62: 505-514.
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005; 65: 10946-10951.
- Liu Y, Han SS, Wu Y, Tuohy TM, Xue H, Cai J, et al. CD44 expression identifies astrocyte-restricted precursor cells. Dev Biol. 2004; 276: 31-46.
- Rebetz J, Tian D, Persson A, Widegren B, Salford LG, Englund E, et al. Glial progenitor-like phenotype in low-grade glioma and enhanced CD133-expression and neuronal lineage differentiation potential in high-grade glioma. PLoS One. 2008; 3: e1936.
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002; 36: 1021-1034.
- Ehtesham M, Yuan X, Kabos P, Chung NH, Liu G, Akasaki Y, et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia. 2004; 6: 287-293.
- Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008; 68: 6043-6048.
- Ueda R, Kohanbash G, Sasaki K, Fujita M, Zhu X, Kastenhuber ER, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc Natl Acad Sci U S A. 2009; 106: 10746-10751.
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004; 64: 7011-7021.
- Inagaki A, Soeda A, Oka N, Kitajima H, Nakagawa J, Motohashi T, et al. Long-term maintenance of brain tumor stem cell properties under at non-adherent and adherent culture conditions. Biochem Biophys Res Commun. 2007; 361: 586-592.
- Soeda A, Inagaki A, Oka N, Ikegame Y, Aoki H, Yoshimura S, et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008; 283: 10958-10966.
- Rich JN, Eyler CE. Cancer stem cells in brain tumor biology. Cold Spring Harb Symp Quant Biol. 2008; 73: 411-420.
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002; 1: 269-277.
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006; 444: 761-765.
- Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, et al. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun. 2007; 360: 553-559.
- Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci U S A. 2002; 99: 14506-14511.
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990; 60: 585-595.
- Berger F, Gay E, Pelletier L, Tropel P, Wion D. Development of gliomas: potential role of asymmetrical cell division of neural stem cells. Lancet Oncol. 2004; 5: 511-514.
- Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006; 441: 1068-1074.
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61: 215-225.
- Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009; 28: 3949-3959.
- Pruszak J, Sonntag KC, Aung MH, Sanchez-Pernaute R, Isacson O. Markers and methods for cell sorting of human embryonic stem cell-derived neural cell populations. Stem Cells. 2007; 25: 2257-2268.
- van Kilsdonk JW, Wilting RH, Bergers M, van Muijen GN, Schalkwijk J, van Kempen LC, et al. Attenuation of melanoma invasion by a secreted variant of activated leukocyte cell adhesion molecule. Cancer Res. 2008; 68: 3671-3679.