
Research Article
Austin Neurosurg Open Access. 2015;2(2): 1032.
Chordoid Meningioma: A Short Series of Five Cases at a Single Institution and Literature Review
Baum J¹, Mrak RE², Bachir S¹ and Medhkour A¹*
1University of Toledo Medical Center, Division of Neurosurgery, USA
2University of Toledo Medical Center, Department of Pathology, USA
*Corresponding author: Medhkour A, Division of Neurosurgery, University of Toledo Medical Center, 3000 Arlington Avenue, Toledo, OH, 43614, USA
Received: June 08, 2015; Accepted: July 21, 2015; Published: July 23, 2015
Abstract
Introduction: The purpose of this study was to report a series of five cases of chordoid meningioma to describe the clinical, pathological, and immunohistochemical features associated with each case and perform a literature review.
Methods: A retrospective chart review was performed on five patients diagnosed with chordoid meningioma and treated at the University of Toledo Medical Center between January 2007 and January 2015. Clinical data obtained includes age, sex, presenting clinical symptoms, duration of symptoms, tumor size, tumor location, treatment, extent of resection, follow-up time, adjuvant therapy, recurrence, and permanent deficits at last follow-up. All cases were diagnosed microscopically by a neuropathologist and immunohistochemistry was performed on two cases.
Results: All patients were female, average age was 55.6 years (range 43- 73), and the most common symptom experienced was headache (n=5). No patients experienced any systemic manifestations of Castleman syndrome. Tumor locations were temporal lobe (n=3), cervical spine (n=1), and frontal lobe (n=1). Average time for follow-up was 38.8 months. Simpson grade I and IV resections were performed in three and two patients, respectively. Recurrence was seen in one patient who underwent grade I resection, and progression was seen in one patient who underwent grade IV resection. No patients underwent adjuvant therapy. Mild deltoid weakness and blurry vision in two patients were the only two deficits noted post resection. Both tumors that had immunohistochemistry performed were positive for progesterone receptor.
Conclusion: Clinical, pathological, and immunohistochemical features of chordoid meningiomas were analyzed and compared with current literature.
Keywords: Brain tumor; Chordoid meningioma
Abbreviations
CT: Computed Tomography; EMA: Epithelial Membrane Antigen; GFAP: Glial Fibrillary Acidic Protein; H&E: Hematoxylin and Eosin; Ki-67 = antigen Ki-67; MRI: Magnetic Resonance Imaging; STR: Subtotal Resection; WHO: World Health Organization
Introduction
The Central Brain Tumor Registry of the United States (www. CBTRUS.org) states meningiomas account for nearly 35% of primary brain tumors, making them the most common primary brain tumor. The World Health Organization (WHO) recognizes 15 variants of meningiomas based on microscopic histology and many of these variants are benign. Chordoid meningioma is a rare atypical variant with an occurrence rate of approximately 0.5% of all meningiomas [1,2]. The chordoid subtype is labeled as a WHO Grade II meningioma because these tumors have increased growth rates, higher recurrence, and greater chances to invade brain parenchyma compared to benign meningiomas (WHO Grade I) [3].
Chordoid meningiomas are very uncommon and, therefore, have limited literature regarding their attributes. The purpose of our study is to highlight characteristics and differences present in these meningiomas as well as the most appropriate treatment approach to prevent recurrence. We report five cases of chordoid meningioma at a single institution to describe the clinical, pathological, and immunohistochemical features associated with each case and perform a literature review.
Methods
This is a retrospective chart review of patients treated at a single institution for chordoid meningioma. All patients presented in this study underwent surgical treatment between January 2007 and November 2014 at the University of Toledo Medical Center. Clinical data obtained includes age, sex, presenting clinical symptoms, duration of symptoms, tumor size, tumor location, treatment, extent of resection, follow-up time, adjuvant therapy, recurrence, and permanent deficits at last follow-up.
Surgical tissue samples were fixed in 10% formalin and embedded in paraffin. These samples were cut in 5-7-μm-thick sections and stained with hematoxylin and eosin per standard protocol. All specimens were reviewed microscopically by one of the authors who is a neuropathologist (REM). Microscopic characteristics, including chordoid features, mitotic features, and necrosis were recorded in the pathologist’s report. Chordoid features were as originally described by Kepes et al., and included cords or nests of vacuolated cells in a mucoid matrix [4].
Immunohistochemistry was performed using standard reagents and techniques using a Ventana Bench Mark automated platform (Ventana Corporation, Tucson AZ). Monoclonal antibodies against the following were used: progesterone receptor (clone 1E2, prediluted), cytokeratin (clones Ae1/Ae3/PK26, prediluted), epithelial membrane antigen (EMA) (clone E29, prediluted), and Ki-67 (clone 30-9, prediluted). All antibodies were obtained from Ventana Medical Systems, Inc., Tucson, AZ. Positive and negative controls were run for each antibody.
Results
Five patients treated for chordoid meningioma were included in our study. Clinical characteristics are shown in Table 1. All patients were female and the average age was 55.6 years (range 43-73 years). The most common symptom experienced was headache (n=5). Other symptoms experienced were gait instability (n=2), visual disturbances (n=2), neck pain (n=1), exophthalmos (n=1), orbital swelling (n=1), and changes in taste (n=1). The duration of symptoms before treatment was sought varied from 3 days to 6 months, with an average time of 2.4 months. No patients experienced any systemic manifestations of Castleman syndrome.
![]()
Case
Sex
Age (years)
Location
Width (cm)
Symptoms
Symptom duration
Simpson grade resection
Adjuvant therapy
Follow-up (months)
Recurrence, progression
Permanent deficits
1
F
73
Right C1-C2
1.5
Headache, neck pain, fall, dizziness
6 months
I
None
54
No
Mild right deltoid weakness
2
F
43
Left sphenoid wing
6.4
Headache, gait disturbance, blurry vision
2 months
I
None
43
Yes
None
3
F
46
Left sphenoid wing
1.6
Headache, left orbital swelling, difficulty writing
3 months
IV
None
42
No
None
4
F
60
Right sphenoid wing
3
Headache, exophthalmos, decreased vision
3 days
IV
None
30
Yes
Mild right eye blurry vision
5
F
56
Left frontal
2.6
Headache
1 month
I
None
25
No
None
F = Female.
Table 1: Clinical features of chordoid meningioma in our case series.
The most common location for the tumors in this study was the temporal lobe (n=3). Two of these tumors were pterional, while the third tumor was so large it encompassed all three divisions of the sphenoid bone (pterional, alar, clinoidal). All three temporal tumors extended into the orbit. The other locations of the last two tumors were frontal lobe and cervical spine (C1-C2). The cervical spine tumor did not extend intracranially and did not involve the brain which makes it an extremely rare occurrence. Average tumor width was 3.0 cm (range 1.5-6.4 cm). Radiologic examples of tumors are demonstrated in Figures 1, 2, and 3.
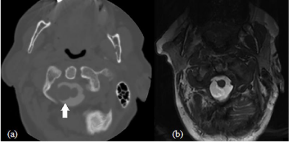
Figure 1: Case 1 radiologic features: (a) Axial cervical spine CT myelogram
showing chordoid meningioma (solid white arrow) abutting the right side of
the cervical spinal cord at C1-C2 (b) T2 axial cervical spine MRI post mass
resection.
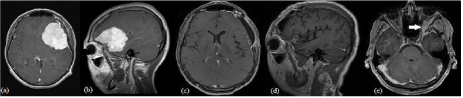
Figure 2: Case 2 MRI features: Chordoid meningioma encompassing parts of the left frontal and temporal lobes and showing midline shift in (a) T1 with contrast
axial imaging and (b) T1 with contrast sagittal imaging. One month post Simpson grade I resection of mass in (c) T1 with contrast axial imaging and (d) T1 with
contrast sagittal imaging. (e) T1 with contrast axial imaging of meningioma recurrence demonstrating an enhancing lesion located along the medial aspect of the
left great sphenoid wing with extension into the lateral orbit (solid white arrow).
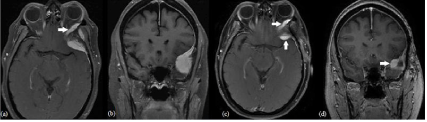
Figure 3: Case 3 MRI features: Chordoid meningioma involving the left sphenoid bone and temporal fossa with extension into the left orbit (solid white arrow) in
(a) T1 axial with contrast and (b) T1 coronal with contrast. Residual tumor after Simpson grade IV resection (solid white arrows) in (c) T1 axial with contrast and
(d) T1 coronal with contrast.
Follow-up data was available for all five patients. Average time for follow-up was 38.8 months (range 25-54 months). Total resection was performed in three patients, one of whom experienced a tumor recurrence at 5 months post resection. None of the three patients underwent adjuvant therapy or additional surgical resections. Subtotal resection was performed in two patients. One of these patients had progression of tumor size at 15 months post resection, and subsequently underwent additional surgical resections. None of these patients underwent adjuvant therapy and no deaths were recorded in our study. Deficits post-resection were found in two patients, and both had very mild symptoms. The symptoms involved deltoid weakness in case 1 and blurry vision in the right eye for case 4. A comparison of clinical features for our study and several other case series is listed in Table 2.
![]()
Kepes et al.[4]
Couce et al.[2]
Tena-Suck et al.[7]
Lin et al.[9]
Zhu et al.[6]
Wang et al.[8]
Epari et al.[10]
Jee et al.[11]
Passacantilli et al.[12]
Our study
Number of cases
7
42
10
11
17
30
12
16
7
5
Mean age (years)
14.8
47.4
42.4
60.8
50.9
49.5
32.4
40
53.9
55.6
Age range (years)
8-19
12-77
35-52
43-77
17-74
16-74
12-67
14-74
29-72
43-73
Number of patients age ≤ 18 years
6 (85.7%)
2 (5.2%)
None
None
1 (5.9%)
1 (3.3%)
3 (25%)
4 (25%)
None
None
Sex ratio (male:female)
1:1.3
1:1
1:2.3
1:0.83
1:1.1
1:1.3
1:1.4
1:1.7
1:0.4
All female
Simpson grading used
No
No
No
No
No
Yes
No
Yes
Yes
Yes
Cases with STR at first surgery
Not described
13 (31%)
None
2 (18%)
2 (11.8%)
4 (13.3%)
1 (8.3%)
None
1 (14.3%)
2 (40%)
Cases with adjuvant therapy
Not described
Not described
7 (70%)
None
5 (29.4%)
9 (30%)
None
8 (50%)
2 (28.6%)
None
Follow-up period
6mo – 5yrs
1mo – 16yrs
2.3yrs – 5.6yrs
1mo – 17yrs
1yr – 5.8yrs
1yr – 14.8yrs
3mo – 2yrs
3mo – 11.3yrs
2.9yrs – 13.3 yrs
2yrs – 4.5yrs
Recurrence, progression
2 (28.7%)
14 (39%)
7 (70%)
2 (18%)
1 (6%)
5 (16.7%)
None
6 (37.5%)
2 (28.6%)
2 (40%)
Mean time to recurrence post-op
3.7 yrs
5.6 yrs
Not described
10.4 yrs
2.6 yrs
2.7 yrs
Not described
3.8 yrs
3.5 yrs
10 mo
STR = Subtotal Resection.
Table 2: Comparison of chordoid meningioma clinical features in various case series.
Mitotic figures were identified in one case (case 1). Necrosis, small cells, nuclear atypia, and inflammation were not seen in any surgical specimen. All patients were diagnosed with chordoid meningioma based on the presence of chordoid features. Immunohistochemistry was done on two surgical specimens (case 2, case 3). Both chordoid meningiomas were positive for progesterone receptors and negative for EMA. One meningioma was positive for cytokeratin and one was negative. Ki-67 immunolabeling was done on one specimen and the tumor labeling rate was estimated to be approximately 10%. Histology examples are demonstrated in Figures 4 and 5.
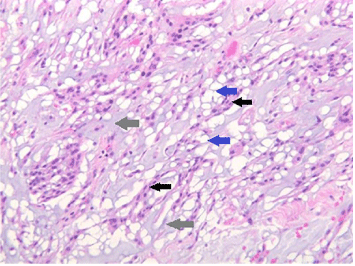
Figure 4: Chordoid features showing epithelioid cells with a vacuolated
appearance (solid blue arrows) and small uniform nuclei arranged in nests
and cords (solid black arrows) against a mucoid matrix (solid gray arrows)
(H&E x 200).
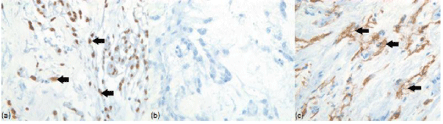
Figure 5: Immunohistochemistry demonstrations: Tumor cells show nuclear immunoreactivity for progesterone receptor (solid black arrows) in (a) case 2 (x200).
Tumor cells were immunonegative for cytokeratin in (b) case 2, but immunopositive (solid black arrows) in (c) case 3 (x200).
Discussion
The first case of chordoid meningioma was originally reported by Connors et al in 1980, who described a 15-year old male diagnosed with a brain tumor and suffering from short stature and sexual development arrest [5]. The patient also had signs of Castle man syndrome, a systemic inflammatory response characterized by hypochromic microcytic anemia unresponsive to iron therapy, dysgammaglobulinemia, and/or hepatosplenomegaly. Diagnosis at the time was uncertain, with consideration of both chordoma and angioblastic meningioma. This case, along with six other cases, was later reviewed in 1988 by Kepes et al., who coined the term “chordoid” meningioma to describe the histology as appearing similar to chordoma (clusters of cells containing intracytoplasmic vacuoles against a mucoid background) but with a gross appearance and location consistent with meningioma [4].
Chordoid meningiomas have been associated with Castleman syndrome since the publication of the first case series in 1988, in which all patients had these signs. The lack of systemic inflammatory signs in subsequent studies after Kepes et al. lead to the belief that Castleman syndrome may be preferentially seen in children since the original study was done on seven patients aged 8-19 years and eight subsequent studies were done on 145 patients with an average age of 47.2 years (range 12-77 years) [2,6-12]. However, aside from the seven patients described by Kepes et al., only twelve other cases associated with Castleman syndrome have been published to date, despite over 200 cases of published chordoid meningiomas [6,13- 21]. The average age of these twelve patients is 32 years (range 15-55 years), with six of the twelve patients being over 35 years, and four of the twelve patients being over 45 years. Castleman syndrome is not found only in children, but these statistics give credence to the belief that this syndrome has a predilection for younger patients.
The largest case series, to date, of chordoid meningioma patients found a 1:1 male to female ratio [2]. However, there have been several other case series showing a female predilection, with male to female ratios ranging from 1:1.3 to 1:2.3 [7,8,10,11]. Coincidentally, all five of our patients were female. Chordoid meningioma mainly occurs in supratentorial areas similar to meningiomas in general. In large studies done on chordoid meningiomas, supratentorial locations were seen in 101 of the 122 cases (82.8%) [2,6-10]. Our results show 80% of our patients had supratentorial locations, which is in agreement to the literature. Unusual sites for chordoid meningiomas include the orbits, jugular foramen, cervical spine, the ventricular system, intraparenchymal, and lung [22-29]. One of our patients had a cervical spine chordoid meningioma located at C1-C2, and only two other cases have been reported to date of cervical spine chordoid meningioma [2, 25].
The most common presenting symptom of chordoid meningioma in six of the largest case series to date is headache [2,6-9,11]; most likely due to increased intracranial pressure, but symptoms largely vary from patient to patient depending on tumor location and size. Other common symptoms noted in the case series include visual and mental changes, seizures, weakness, dizziness, gait changes, and hemiparesis. Exophthalmos and periorbital swelling were seen in two of our five patients. Each of these patients had tumor extension into the orbit exhibiting the symptoms.
The Simpson grading scale for meningioma resection was first described by Simpson in 1957 [30]. In a study of 391 patients with WHO grade I meningioma, further treatment after initial resection was needed approximately 5 times more often for grade II and III resection patients versus grade I resection patients, and 13 times more often for grade IV and V resection patients versus grade I resection patients [31]. Complete removal of the meningioma, including all dural and bony infiltrations, is the ultimate goal of therapy, but grade I resection may be difficult in certain locations such as the cavernous sinus, suprasellar region, petroclival region, and near the optic nerve. Two patients in this study had Simpson grade IV resection due to the tumor location near the optic nerve.
Chordoid meningioma is designated as a WHO grade II meningioma and has higher recurrence rates compared to benign (WHO grade I) meningiomas. In a series of 581 patients diagnosed with benign meningiomas who underwent resection, 5-year recurrence rates were 12% post total resection and progression rates were 39% post subtotal resection [32]. In a review of 8 chordoid meningioma case series involving 145 patients, gross total resection was performed in 123 patients and subtotal resection in 22 patients, and recurrence was seen in 14% of patients who underwent gross total resection versus an 82% progression rate for subtotal resection patients [2,6- 12]. Unfortunately, Simpson grading was reported in only 3 of the 8 series. In our patients subtotal resection (Simpson grade 4 or higher) was performed in 2 cases and progression was seen in one of these patients. Recurrence was seen in 1 of the 3 patients who underwent Simpson grade I resection (case 2). This patient did have primary tumor extension into the orbit and the neurosurgeon who performed the resection did not rule out the possibility of residual tumor in the orbit post resection which could explain the recurrence even though no residual tumor was found on follow up MRI imaging one month after initial resection. The stark differences in recurrence between subtotal and total resection in these series of chordoid meningioma patients emphasizes the importance of complete resection during the initial surgery.
Adjuvant radiotherapy continues to be a practiced method in treating WHO grade II meningiomas and many retrospective studies have been published debating its effectiveness. Pearson et al documented that rates of adjuvant radiotherapy in subtotally resected WHO grade II meningiomas nearly doubled over the years [33].There have been mixed results in terms of adjuvant radiotherapy use post subtotal resection of WHO grade II meningiomas, with some studies showing improved outcome [34] and others showing no significant change in outcome [35,36]. Results seem more one sided in regards to using adjuvant radiotherapy post gross total resection of WHO grade II meningiomas, with several studies showing no overall significant effect on outcome [35-38] or recurrence rates [39]. Furthermore, Table 2 shows the number of chordoid meningioma patients in each case series that underwent adjuvant therapy which varied from 0-70% of patients. Recurrence or progression of tumor growth was seen in some series where adjuvant therapy was aggressively used, and lack of recurrence was seen in some series where adjuvant therapy was used less often. These studies have all been retrospective, and the use of adjuvant radiotherapy for WHO grade II meningiomas, and more specifically chordoid meningiomas, will likely continue to be an area of debate until prospective studies are published.
Most meningiomas can be diagnosed radiologically with Magnetic Resonance Imaging (MRI), but subtyping a meningioma is done histologically. Chordoid meningioma contains groups of vacuolated spindle or epithelioid cells fixed in a mucoid stroma [12,40] that may also contain inflammatory cells [2,41]. These histological characteristics have overlapping qualities with several other tumors. The differential diagnosis includes myxopapillary ependymoma, chordoid glioma, chordoma, metastatic mucinous carcinoma, chordoid sarcoma, low-grade chondrosarcoma, enchondroma, and myxoidchondrosarcoma [2,29,42]. Immunological staining is useful in establishing a correct diagnosis. In a series of 42 chordoid meningioma cases, all tumors were shown to be immunoreactive for vimentin and at least focal immunoreactivity to EMA, and nonreactive for GFAP [2]. A panel of EMA, GFAP, D2-40, and pancytokeratin was demonstrated to be useful in analyzing histological mimics, with chordoid meningioma being immunopositive for EMA in all 4 chordoid meningioma cases, positive for D2-40 in half of the cases, and nonreactive to pan-cytokeratin and GFAP in all cases [43]. Another study found D2-40, EMA, brachyury, and GFAP to be the most useful panel, with chordoid meningioma being immunopositive for EMA in 90% of the cases and D2-40 in 80% of the cases, and nonreactive for brachyury and GFAP in 100% of the cases [42]. This study also found 20% of cases to test positive for pankeratin [42]. Both chordoid meningiomas in our study that were analyzed using immunohistochemistry were negative for EMA despite its strong association with this type of tumor. Furthermore, both cases were also positive for progesterone receptor, and one case was positive for cytokeratin despite the lack of association with chordoid meningioma.
Progesterone receptors have been found to occur in approximately 45% of WHO grade II meningiomas in both men and women [44]. Anti-progesterone therapy using mifepristone (RU 486) has been shown by several studies and also by an unpublished study performed by the senior author of this manuscript (AM) to have some success in either arresting tumor progression or decreasing volume of benign meningiomas [45-47]. The mechanism of action may not be straight forward, as Matsuda et al. have indicated that anti-progesterone therapy may exert its effect on meningiomas through glucocorticoid receptors or some other pathway besides progesterone receptors [48]. Aside from the poorly understood mechanism, this therapy has not been largely accepted and, moreover, its use in treating WHO grade II meningiomas has yet to be reviewed. Other interesting hormonal modulation therapies for meningiomas include growth hormone receptor antagonists and somatostatin agonists [49-51]. Unfortunately, only small reports are available for human studies regarding these therapies, and no objective improvements were found [50, 51].
Conclusion
Chordoid meningioma is an uncommon subtype of meningiomas and exhibits an increased risk of recurrence, especially when subtotally resected. We report 5 cases of chordoid meningioma at a single institution arising in the cervical spine, frontal lobe, and sphenoid wing. None of our patients had any symptoms of Castleman syndrome. Chordoid meningiomas may present a great challenge if large and infiltrating important structures. Subtotal resection (Simpson grade 4) was performed in two patients due to the proximity to the optic nerve. Adjuvant radiotherapy was not performed in any of the five cases. Microscopically, all tumors exhibited characteristic chordoid morphology. Two of the five cases had immunochemical evaluation, and both cases were immunoreactive for progesterone receptor. The best therapeutic approach for chordoid meningiomas is radical resection during the first surgery with every effort being made to leave no residual that may lead to recurrence. Radiation therapy may be utilized judiciously although it is not devoid of complications and effectiveness has not been completely established.
References
- Di Ieva A, Laiq S, Nejad R, Schmitz EM, Fathalla H, Karamchandani J, et al. Chordoid meningiomas: incidence and clinicopathological features of a case series over 18 years. Neuropathology. 2015; 35: 137-147.
- Couce ME, Aker FV, Scheithauer BW. Chordoid meningioma: a clinicopathologic study of 42 cases. Am J Surg Pathol. 2000; 24: 899-905.
- Rychlŷ B, Sidlovã H, Daniă D. [The 2007 World Health Organisation classification of tumours of the central nervous system, comparison with 2000 classification]. Cesk Patol. 2008; 44: 35-36.
- Kepes JJ, Chen WY, Connors MH, Vogel FS. "Chordoid" meningeal tumors in young individuals with peritumoral lymphoplasmacellular infiltrates causing systemic manifestations of the Castleman syndrome. A report of seven cases. Cancer. 1988; 62: 391-406.
- Connors MH. Growth and maturation arrest, hypochromic anemia and hyperglobulinemia associated with a brain tumor. West J Med. 1980; 133: 160-163.
- Zhu HD, Chen H, Xie Q, Gong Y, Mao Y, Zhong P, et al. Chordoid meningioma: a retrospective study of 17 cases at a single institution. Chin Med J (Engl). 2013; 126: 789-791.
- Tena-Suck ML, Collado-Ortìz MA, Salinas-Lara C, García-López R, Gelista N, Rembao-Bojorquez D. Chordoid meningioma: a report of ten cases. J Neurooncol. 2010; 99: 41-48.
- Wang XQ, Mei GH, Zhao L, Li ST, Gong Y, Zhong J, et al. Clinical features and treatment of intracranial chordoid meningioma: a report of 30 cases. Histopathology. 2013; 62: 1002-1017.
- Lin JW, Ho JT, Lin YJ, Wu YT. Chordoid meningioma: a clinicopathologic study of 11 cases at a single institution. Journal of neuro-oncology. 2010; 100: 465-473.
- Epari S, Sharma MC, Sarkar C, Garg A, Gupta A, Mehta VS. Chordoid meningioma, an uncommon variant of meningioma: a clinicopathologic study of 12 cases. J Neurooncol. 2006; 78: 263-269.
- Jee TK, Jo KI, Seol HJ, Kong DS, Lee JI, Shin HJ. Clinical features and treatment outcome of chordoid meningiomas in a single institute. J Korean Neurosurg Soc. 2014; 56: 194-199.
- Passacantilli E, Lapadula G, Caporlingua F, Lenzi J, Antonelli M, Santoro F, et al. Chordoid meningioma: a retrospective series of seven consecutive cases. Neurol Sci. 2013; 34: 1985-1989.
- Civit T, Baylac F, Taillandier L, Auque J, Hepner H. [Chordoid meningiomas. Clinical, neuroradiological and anatomopathological aspects. Apropos of a new case and review of the literature]. Neurochirurgie. 1997; 43: 308-313.
- Kobata H, Kondo A, Iwasaki K, Kusaka H, Ito H, Sawada S. Chordoid meningioma in a child. Case report. J Neurosurg. 1998; 88: 319-323.
- Arima T, Natsume A, Hatano H, Nakahara N, Fujita M, Ishii D, et al. Intraventricular chordoid meningioma presenting with Castleman disease due to overproduction of interleukin-6. Case report. J Neurosurg. 2005; 102: 733-737.
- Kaloshi G, Antonelli M, Vreto G, Lame A, Kerri I, Bushati T, et al. Report of two cases of chordoid meningioma in patients with Castleman syndrome. J Neurooncol. 2011; 104: 395-397.
- Lee DK, Kim DG, Choe G, Chi JG, Jung HW. Chordoid meningioma with polyclonal gammopathy. Case report. J Neurosurg. 2001; 94: 122-126.
- Yano H, Shinoda J, Hara A, Shimokawa K, Sakai N. Chordoid meningioma. Brain Tumor Pathol. 2000; 17: 153-157.
- Denaro L, Di Rocco F, Gessi M, Lauriola L, Lauretti L, Pallini R, et al. Pyrogenic cytokine interleukin-6 expression by a chordoid meningioma in an adult with a systemic inflammatory syndrome. Case report and review of the literature. J Neurosurg. 2005; 103: 555-558.
- Hamels M, Mariman A, Kalala O, Van den Broecke C, Delesie L, Tobback E, et al. Chordoid meningioma in an adult patient presenting with chronic fatigue and systemic inflammation. Acta Clin Belg. 2013; 68: 444-448.
- Jeon CJ, Kim MJ, Lee JS, Lee JH, Kong DS, Shin HJ, et al. Castleman's disease associated with a cerebellar chordoid meningioma and intestinal lymphangiectasia. Child's nervous system: ChNS: official journal of the International Society for Pediatric Neurosurgery. 2010; 26: 1647-1652.
- Salinero E, Beltran L, Costa JR. Intraoperative cytologic diagnosis of chordoid meningioma. A case report. Acta Cytol. 2004; 48: 259-263.
- Rowsell C, Sirbovan J, Rosenblum MK, Perez-Ordoñez B. Primary chordoid meningioma of lung. Virchows Arch. 2005; 446: 333-337.
- Takei H, Rivera A, Suzuki H, Bahrami A, Powell SZ. Jugular foramen chordoid meningioma. Pathol Int. 2006; 56: 397-401.
- Ibrahim A, Galloway M, Leung C, Revesz T, Crockard A. Cervical spine chordoid meningioma. Case report. J Neurosurg Spine. 2005; 2: 195-198.
- Song KS, Park SH, Cho BK, Wang KC, Phi JH, Kim SK. Third ventricular chordoid meningioma in a child. J Neurosurg Pediatr. 2008; 2: 269-272.
- Nambiar A, Pillai A, Parmar C, Panikar D. Intraventricular chordoid meningioma in a child: fever of unknown origin, clinical course, and response to treatment. Journal of neurosurgery Pediatrics. 2012; 10: 478-481.
- Wind JJ, Jones RV, Roberti F. Fourth ventricular chordoid meningioma. J Clin Neurosci. 2010; 17: 1301-1303.
- Xi S, Zhang Y, Lin S, Liang J, Zeng J, Wu Q. Intraparenchymal chordoid meningioma: a case report and review of the literature. Int J Surg Pathol. 2012; 20: 600-605.
- Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957; 20: 22-39.
- Hasseleid BF, Meling TR, Rønning P, Scheie D, Helseth E. Surgery for convexity meningioma: Simpson Grade I resection as the goal: clinical article. J Neurosurg. 2012; 117: 999-1006.
- Stafford SL, Perry A, Suman VJ, Meyer FB, Scheithauer BW, Lohse CM, et al. Primarily resected meningiomas: outcome and prognostic factors in 581 Mayo Clinic patients, 1978 through 1988. Mayo Clin Proc. 1998; 73: 936-942.
- Pearson BE, Markert JM, Fisher WS, Guthrie BL, Fiveash JB, Palmer CA, et al. Hitting a moving target: evolution of a treatment paradigm for atypical meningiomas amid changing diagnostic criteria. Neurosurgical focus. 2008; 24: E3.
- Mair R, Morris K, Scott I, Carroll TA. Radiotherapy for atypical meningiomas. J Neurosurg. 2011; 115: 811-819.
- Lee KD, DePowell JJ, Air EL, Dwivedi AK, Kendler A, McPherson CM. Atypical meningiomas: is postoperative radiotherapy indicated? Neurosurg Focus. 2013; 35: E15.
- Detti B, Scoccianti S, Di Cataldo V, Monteleone E, Cipressi S, Bordi L, et al. Atypical and malignant meningioma: outcome and prognostic factors in 68 irradiated patients. J Neurooncol. 2013; 115: 421-427.
- Aizer AA, Arvold ND, Catalano P, Claus EB, Golby AJ, Johnson MD, et al. Adjuvant radiation therapy, local recurrence, and the need for salvage therapy in atypical meningioma. Neuro Oncol. 2014; 16: 1547-1553.
- Komotar RJ, Iorgulescu JB, Raper DM, Holland EC, Beal K, Bilsky MH, et al. The role of radiotherapy following gross-total resection of atypical meningiomas. J Neurosurg. 2012; 117: 679-686.
- Aghi MK, Carter BS, Cosgrove GR, Ojemann RG, Amin-Hanjani S, Martuza RL, et al. Long-term recurrence rates of atypical meningiomas after gross total resection with or without postoperative adjuvant radiation. Neurosurgery. 2009; 64: 56-60.
- Lee KH, Lall RR, Chandler JP, Bigio EH, Mao Q. Pineal chordoid meningioma complicated by repetitive hemorrhage during pregnancy: case report and literature review. Neuropathology: official journal of the Japanese Society of Neuropathology. 2013; 33:192-198.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114: 97-109.
- Sangoi AR, Dulai MS, Beck AH, Brat DJ, Vogel H. Distinguishing chordoid meningiomas from their histologic mimics: an immunohistochemical evaluation. Am J Surg Pathol. 2009; 33: 669-681.
- Cho HY, Lee M, Takei H, Dancer J, Ro JY, Zhai QJ. Immunohistochemical comparison of chordoma with chondrosarcoma, myxopapillary ependymoma, and chordoid meningioma. Applied immunohistochemistry & molecular morphology: AIMM / official publication of the Society for Applied Immunohistochemistry. 2009; 17: 131-138.
- Roser F, Nakamura M, Bellinzona M, Rosahl SK, Ostertag H, Samii M. The prognostic value of progesterone receptor status in meningiomas. J Clin Pathol. 2004; 57: 1033-1037.
- Grunberg SM, Weiss MH, Spitz IM, Ahmadi J, Sadun A, Russell CA, et al. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J Neurosurg. 1991; 74: 861-866.
- Touat M, Lombardi G, Farina P, Kalamarides M, Sanson M. Successful treatment of multiple intracranial meningiomas with the antiprogesterone receptor agent mifepristone (RU486). Acta Neurochir (Wien). 2014; 156: 1831-1835.
- Lamberts SW, Tanghe HL, Avezaat CJ, Braakman R, Wijngaarde R, Koper JW, et al. Mifepristone (RU 486) treatment of meningiomas. J Neurol Neurosurg Psychiatry. 1992; 55: 486-490.
- Matsuda Y, Kawamoto K, Kiya K, Kurisu K, Sugiyama K, Uozumi T. Antitumor effects of antiprogesterones on human meningioma cells in vitro and in vivo. J Neurosurg. 1994; 80: 527-534.
- McCutcheon IE, Flyvbjerg A, Hill H, Li J, Bennett WF, Scarlett JA, et al. Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude mice. J Neurosurg. 2001; 94: 487-492.
- García-Luna PP, Relimpio F, Pumar A, Pereira JL, Leal-Cerro A, Trujillo F, et al. Clinical use of octreotide in unresectable meningiomas. A report of three cases. J Neurosurg Sci. 1993; 37: 237-241.
- Drake WM, Grossman AB, Hutson RK. Effect of treatment with pegvisomant on meningioma growth in vivo. Eur J Endocrinol. 2005; 152: 161-162.