
Special Article - Anemia
Ann Nutr Disord & Ther. 2021; 8(1): 1067.
Prevalence of Sickle Cell Anemia in Association of Plasmodium falciparum from Rajnandgaon, India
Tripathi S¹, Mishra N² and Kumar A²*
¹Department of Science and Technology, Women Scientist-A, India
²Department of Biotechnology, Government VYT PG Autonomous College, India
*Corresponding author: Anil Kumar, Department of Biotechnology, Government VYT PG Autonomous College, Durg-491001, Chhattisgarh, India
Received: December 15, 2020; Accepted: January 07, 2021; Published: January 14, 2021
Abstract
Sickle cell anemia is a blood disorder resulting from the inheritance of abnormal genes from parents. It is caused due to mutation in the β-globulin gene. Sickle cell anemia is widespread across the world and in Central India. The present study was undertaken to study the prevalence of the disorder in the Rajnandgaon district of Central India. A random sampling of 6088 people was done to test the sickle cell anemia problem by slide test method and a total of 249 (4.09%) people were found sickled positive. Further electrophoresis test was performed for all 249 of which 67 were found homozygous (HbSS) and 182 were found heterozygous (HbAS) positive.
Besides the above analysis, chloroquine prophylaxis associated with a high prevalence of Plasmodium falciparum Pfcrt K76T mutation in people (n=26) with sickle cell anemia was also analyzed. The genotype of the subject was screened using the hemoglobin electrophoresis system and the P. falciparum Pfcrt genotyping was carried out using PCR-Restriction Fragment Length Polymorphism (RFLP). The prevalence rate of Pfcrt K76T mutant gene was proportionately found higher in the hemoglobin SS (n=40, m=32, r=0.67) genotype individuals than the hemoglobin AS (n=52, m= 27, r=0.519) and AA (n=182, m= 68, r= 0.37).
Keywords: Sickle cell anemia; P. falciparum; Chloroquine; Pfcrt K76T, PCR-RFLP
Introduction
Sickle Cell Hemoglobin (HbS) is the first molecular disease known to man [1]. It is a structural variant of hemoglobin in which glutamic acid (an amino acid), at position no.6 of a β-globin chain of hemoglobin is replaced by valine. This happens due to the change of nucleotide, adenine to thymine (GAG/GTG) of codon 6 of a β-globin gene, located on the short arm of chromosome 11. This substitution of amino acid changes the net charge of hemoglobin, oxygen affinity, and three-dimensional structure of hemoglobin, thus rendering it unstable hemoglobin. Sickle hemoglobin gets polymerized at low oxygen tension and deforms the red cell from discoid shape to sicklelike form causing a lot of pathogenicities [2]. Due to its genetic nature of origin, the disease is inheritable following the Mendelian Principle. In malaria-infested areas, the high frequency of hemoglobinopathies, such as Sickle-Cell Disease (SCD), exhibits their protective role against P. falciparum malaria [3]. However, in homozygous sickle hemoglobin (SS) cases, the tenacity of P. falciparum could set off acute hemolytic events [4] and/or recurrence of Vaso-Occlusive Crises (VOCs).
The exact mechanism by which sickle-cell trait (AS) condition imparts resistance to malaria is not known. Various factors are likely to be involved that contribute to varying degrees of defense against malaria [5]. Erythrocytes of sickle cell trait, infected with the P. falciparum parasite are deformed since the parasite drastically decreases the oxygen tension within the red cells as it carries out its metabolism also. This deformation of sickle trait (AS) erythrocytes makes these cells abnormal and obvious targets for phagocytosis [6].
Plasmodium genus is approximated to have evolved around 150 million years ago, even before the evolution of Homo sapiens [7]. So, humans have evolved in the presence of malaria, and this co-evolution has shaped the human genome [8]. Various polymorphisms are protective against severe forms of malaria. The worldwide distributions of Hemoglobinopathies like sickle cell anemia, thalassemias, Glucose-6-phosphate dehydrogenase (G6PD) deficiency, and blood group polymorphisms suggests the prevalence of malaria, indicating that malaria has been a selective force for such mutations [9][7]. Also, genetic variations in human P450 genes (CYP2C8, CYP3A4, and CYP2A6) results in differential metabolism of anti-malarial drugs in humans and have important imputations in both anti-malarial drug efficacy and bearability [10,11].
Chloroquine is a 4-amino-quinoline anti-malarial drug that interferes with the sequestration of toxic heme, which is produced when hemoglobin is digested by an intra-blood parasite to obtain essential amino acids. The parasite crystallizes heme into hemozoin in its acidic digestive vacuoles.
Chloroquine (and other similar drugs) binds to heme and prevents the detoxification process [12]. Parasite resistance is believed to be achieved by the decreased accumulation of chloroquine in the digestive vacuole of the parasite.
In 2000, the P. falciparum chloroquine resistance transporter (pfcrt), the key gene involved in this resistance, was discovered [13]. CQ resistance is associated with a T76 mutation of the P. falciparum chloroquine resistance transporter gene (Pfcrt) [13] while a multidrug resistance analog (Pfmdr1) Y86 variation may modulate its degree [14]. The gene is present on chromosome 7 and encodes a drug and metabolite transporter protein (PfCRT) located on the membrane of the digestive vacuole. A unique mutation pfcrt K76T, was found in connection with other compensatory amino acid residue changes at loci PfCRT 72-76, where PfCRT 72-76 CVMNK is the sensitive haplotype and CVIET and SVMNT are considered the most common resistant haplotypes [15].
Chloroquine resistance depends upon the genetic background of the parasite line i.e., pfcrt K76T enhances the tolerance to chloroquine so that resurgence is likely to occur, but it does not always result in clinical dereliction [12].
According to a hypothesis regarding the resistance is the protonation of chloroquine in the acidic environment of the digestive vacuole. Efflux of the chloroquine (CQ2+) is reduced by the charged lysine (PfCRT K76) amino acid residue in the Chloroquine sensitive strain. When lysine is replaced by neutral amino acid residue threonine (PfCRT 76T) then CQ2+ can egresses down its concentration gradient through PfCRT, eliminating the drug from its target site. Another drug, Verapamil, which is a calcium (Ca2+) channel blocker, is can reverse this resistance by competing with chloroquine for binding at PfCRT, thereby blocking the efflux of the drug from the digestive vacuole.
For a very long time, it is supposed that the malarial parasite has tempted RBC for various mutations for protection. Probably sickling is one of them. A lot of Epidemiological and biochemical evidence is available related to the concept. Chhattisgarh state (India) is one of the major centers of malaria epidemics, especially for Plasmodium falciparum. We have noticed the development of malaria among the sickled population against the previous concept.
So banking on the prevalence of both malaria epidemics and sickling in the society of Chhattisgarh state (India), we have undertaken the present study to detect mutation in Plasmodium falciparum in an association of Heterozygous Sickling (HbAS) and Homozygous Sickling (HbSS) with a correlation of normal hemoglobin.
Material and Method
For the study, seven villages of Rajnandgaon District of Chhattisgarh state (India) within a radius of 60-70kms from district headquarters were selected. Two milliliters of intravenous blood was collected from each person by paramedical staff following ICMR and Institutional Ethics Committee norms. From the first village total, 6088 samples were collected from all seven villages (Table 1). During sample collection, it was taken into note that some of the donors were suffering from malaria and had taken chloroquine as medicine.
![]()
S.No.
Village
Total Sample
Sickle Positive after Slide Test
Percentage
(%)
Electrophoresis Test
Homozygous
(SS)
Heterozygous
(AS)
1.
Village First
982
52
5.29%
18
34
2.
Village Second
767
19
2.47%
6
13
3.
Village Third
1012
58
5.73%
11
47
4.
Village Fourth
894
43
4.80%
9
34
5.
Village Fifth
759
24
3.16%
6
18
6
Village Sixth
852
33
3.87%
12
21
7.
Village Seventh
822
20
2.43%
5
15
Total
6088
249
4.09%
67 (26.90%)
182 (73.09%)
Table 1: Prevalence of Sickle cell Anemia with a homozygous and heterozygous variation.
After sample collection primary screening was done by the slide test method, followed by electrophoresis in Starch-Agarose Gel electrophoresis for confirmation and hemoglobin type was determined by comparing with the standard. Blood from those persons who were suffering from sickling with malaria and malaria without sickling was blotted on a piece of blotting paper. DNA of Plasmodium falciparum was extracted from dried blood spots following the modified Saponin Chelex method. Genotyping of the resistance marker gene Pfcrt K76T was carried out by PCR - RFLP. Two primers viz. 5’AATTAAAgTTgAgTTTCggA3’ and 5’TgTgCTCATgTgTTTAAACTT3’ (Bgenei) for amplification of Pfcrt sequences were used, following amplification of the Pfcrt sequence restriction digestion of the product was carried out by restriction endonuclease Apo I (Bgenei). The product was observed in 2% agarose gel on electrophoresis, stained with EtBr, and examined in Gel Documentation System.
Result
The study showed that out of a total of 6088 samples collected, 249 were found sickle positive (4.09%). From the first village out of 982, 52 were found sickle positive (18-SS and 34-AS ); from the second village out of 767, 19 were found sickle positive ( 6-SS and 13- AS ); from the third village out of 1012, 58 were found sickle positive ( 11-SS and 47-AS ); from the fourth village out of 894, 43 were found sickle positive (9-SS and 34-AS); from the fifth village, out of 759, 24 were found sickle positive (6-SS and 18-AS); from the sixth village, out of 852, 33 were found sickle positive (12-SS and 21-AS) and from the seventh village, out of 822 samples collected, 20 were found sickle positive (5-SS and 15-AS). Out of a total of 6088 samples, 274 (4.38%) persons were found infected with malaria parasites (Table 2). Following amplification of the Pfcrt fragments of codon 76 and restriction digestion of the amplified products with Apo I, we found the prevalence rate of mutant genes and the prevalence rate of Pfcrt K76T mutant gene was reported higher in hemoglobin SS (n=40, m=32, r=0.67) genotype individuals than the hemoglobin AS (n=52, m=27, r=0.519) and normal hemoglobin AA (n=182, m=68, r=0.37) subjects (Table 2, Figure1). This indicates a kind of resistance in term of the proliferation of parasites occurred in the hemoglobin AS and SS individuals. It was found that there is a significant difference (p<0.05) in the prevalence rate of mutant Pfcrt genes between hemoglobin SS, AA, and AS.
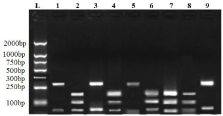
Figure 1: Agarose gel depicting the K76 and 76T after RFLP digestion with
Apo I: Lane 1 is DNA Molecular Weight Ladder (100bp), Lane 2,4,6,7 & 8
showing the K76 (Wild type) bands (with 137,124 and 10bp bands); Lanes
1, 3, 5 & 9 showing the 76T (mutant) bands (with 260bp and 10bp bands).
![]()
S. No.
Total Malaria patients & (%)
Patients with normal genotype (AA)
Patients with homozygous genotype (SS)
Patients with heterozygous genotype (AS)
No. of Patients
Patients with Mutant (76T)
No. of Patients
Patients with Mutant (76T)
No. of Patients
Patients Mutant (76T)
1
274 (4.38%)
182
68
40
27
52
21
Table 2: Prevalence of Pfcrt K76T mutation in P. falciparum among Hb AA, HbSS and HbAS.
Discussion
Our finding indicates that maximum resistance in terms of proliferation of parasites occurred in hemoglobin SS, but moderate resistance was also reported from hemoglobin AS. However according to an author, genes that select against “severe” falciparum malaria include hemoglobin S gene, Thalassemia gene, Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency gene, Ovulocytosis, and Duffy gene, and protection is afforded only to heterozygous individuals (hemoglobin AS) in severe malaria [16]. Several factors have been earmarked to contribute to the emergence of these resistant genes notably environmental factors, host factors, and drug pressure. This high prevalence in the hemoglobin SS genotyped individuals could be as a result of uncontrolled usage of chloroquine as prophylaxis. There is also a report that chloroquine prophylaxis is responsible for increased drug consumption and increased drug pressure that may lead to the selection of drug-resistant parasites [17].
Shrivastava et al., in 2014 analyzed the correlation of Pfcrt-K76T and Pfmdr1-N86Y mutations with Chloroquine (CQ) resistance in Northeast Indian P. falciparum isolates. They reported that out of 115 P. falciparum isolates, 100 isolates were found to resistant to CQ by the in vitro susceptibility test whereas 15 were found CQ sensitive. They further reported that all the CQ resistant isolates exhibited the presence of Pfmdr1 and Pfcrt mutations. On the other hand, CQ sensitive isolates didn’t exhibit such mutations. They also observed strong linkage disequilibrium between the alleles at Pfmdr1-N86Y and Pfcrt-K76T loci. Their results indicated that Pfmdr1-N86Y and Pfcrt-K76T mutations can be used as molecular markers for the identification of CQ resistance in P. falciparum and recommended the importance of evaluation of CQ in vivo therapeutic efficacy in endemic areas for more effective malaria control strategies [18].
Another study also reported hemoglobin AS resistance against Plasmodium falciparum in comparison to hemoglobin SS [19]. But our findings differ from previous workers we found maximum resistance in hemoglobin SS than AS. Although the mechanism of resistance either in hemoglobin SS and in AS is yet to be worked out, the relevance of the present investigation is that there is an urgent need to control the indiscriminate use of chloroquine and development of substitute therapeutic mechanism to prevent the evolution of resistance gene in Plasmodium falciparum.
Acknowledgement
We acknowledge all the participants who have given samples for study and to the Department of Science and Technology, Government of India, for financial support.
References
- Serjeant GR, Serjeant BE. Sickle Cell Disease. Oxford University. New York Press. 2001.
- Gupta RB. Sickle cell disease load in Madhya Pradesh. RMRCT Update. 2006; 3: 1-5.
- Aidoo M, Terlouw DJ, Kolczak MS. Protective effects of the sickle-cell gene against malaria morbidity and mortality. Lancet. 2002; 359: 1311-1312.
- Bates I, Bedu-Addo G, Bevan DH. Use of immunoglobulin gene rearrangements to show clonal lymphoproliferation in hyper-reactive malarial splenomegaly. Lancet. 1991; 337: 505-507.
- Martin TW, Weisman IM., Zeballos RJ, Stephenson SR. Exercise and hypoxia increase sickling in venous blood from an exercising limb in individuals with sickle-cell trait. Am J Med. 1989; 87: 48-56.
- Luzzatto L, Nwachuku-Jarrett ES, Reddy S. Increased sickling of parasitized erythrocytes as a mechanism of resistance against malaria in the sickle-cell trait. Lancet. 1970; 1: 319-321.
- Evans AG, Wellems TE. Coevolutionary genetics of Plasmodium malaria parasites and their human hosts. Integer. Comp. Biol. 2002; 42: 401-417.
- Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015; 517: 327-332.
- Mohandas N, An X. Malaria and human red blood cells. Med Microbiol Immunol. 2012; 201: 593-598.
- Piedade R, Gil JP. The pharmacogenetics of antimalaria artemisinin combination therapy. Expert Opin Drug Metab Toxicol. 2011; 7: 1185-200.
- Roederer MW, McLeod H,Juliano JJ. Can pharmacogenomics improve malaria drug policy? Bull World Health Organ. 2011; 89: 838-845.
- Muller IB, Hyde JE. Antimalarial drugs: modes of action and mechanisms of parasite resistance. Future Microbiol. 2010; 5: 1857-1873.
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. cell. 2000; 6: 861-871.
- Sanchez C, Lanzer M. Changing ideas on chloroquine in Plasmodium falciparum. Curr Opinion Infect Dis. 2000; 13: 653-658.
- Sibley CH. Understanding drug resistance in malaria parasites: Basic science for public health. Mol Biochem Parasitol. 2014; 195: 107-114.
- Cheesbrough M. Malaria parasites. In: Medical Laboratory Manual for Tropical Countries, v II, II edn. (Low priced EL/BS edition). United Kingdom: Tropical Health Technology. 1992; 221-245.
- Bertin G, Tuikue N, Jafari-Guemouri S. High prevalence of Plasmodium falciparum Pfcrt K76T mutation in pregnant women taking chloroquine prophylaxis in Senegal. Antimicrob Chem.1995; 55: 788-791.
- Shrivastava SK, Gupta RK, Mahanta J, Dubey ML. Correlation of molecular markers, Pfmdr1-N86Y and Pfcrt-K76T, with in vitro chloroquine-resistant Plasmodium falciparum, isolated in the malaria-endemic states of Assam and Arunachal Pradesh, Northeast India. PLoS One. 2014; 9: e103848.
- Tatfeng YM, Agbonlahor DE, Tchounga KS, Omoluc PI, Okoduad M, Yah CS, et al. Chloroquine prophylaxis associated with a high prevalence of Plasmodium falciparum Pfcrt K76T mutation in people with sickle-cell disease in Benin City, Nigeria. J Vector Borne Dis. 2008; 45: 51-55.