
Case Report
Austin J Obstet Gynecol. 2014;1(1): 4.
The Diagnosis and Management of an Adult 46xy Female with Isolated 17, 20-Lyase Deficiency due to a Novel Mutation P. Y35xi Cytochrome B5a Gene
Tracy Wing Yee Yeung1*, Angel on Kei Chan2, Raymond Hang Wun Li1, Chi Chung Shek2, Pak Chung Ho1 and Ernest Hung Yu Ng1
1Department of Obstetrics & Gynecology, The University of Hong Kong, People's Republic of China
2Department of Pathology, Queen Mary Hospital, People's Republic of China
*Corresponding author: Tracy Wing Yee Yeung, Department of Obstetrics and Gynecology, The University of Hong Kong, Queen Mary Hospital, Pokfulam Road, Hong Kong
Received: June 20, 2014; Accepted: July 22, 2014; Published: July 23, 2014
Abstract
Objective: To report the diagnosis and clinical management of an adult Chinese 46 XY woman due to isolated 17,20-lyase deficiency resulting from a homozygous Cytochrome B5A mutation p.Y35X with non-consanguineous parents.
Design: Case Report.
Setting: Gynaecological endocrine clinic in a University Hospital.
Patients: A 29 year-old Chinese woman with 46XY Disorder of Sex Development (DSD) resulting from isolated 17, 20-lyase deficiency and her parents.
Intervention: Investigations with serum hormonal profiles, urine steroid profile, serum methaemoglobin, mutational analyses of the CYP17A1 and CYB5A by polymerase chain reaction (PCR) and DNA sequencing, surgical correction for ambiguous genitalia.
Main outcome measure: Steroid hormones and methaemoglobin levels, urine steroid profile, serum mutational analyses of CYP17A1 and CYB5A genes and post-operative clinical outcomes.
Result: Steroid hormone levels and urine steroid profile suggested isolated 17,20-lyase deficiency with elevated methaemoglobin level. Mutational analysis confirmed a homozygous novel mutation p.Y35X in the CYB5A gene. Correctional surgery restored normal female external genitalia.
Conclusion: Urine steroid profiling is a useful test in pinpointing the exact error of testosterone metabolism and serum methaemoglobin level may be used to screen for abnormal Cytochrome B5 function. Surgical treatment aims at restoring normal external genitalia of the assigned gender.
Keywords: 46XY female; isolated 17,20-lyase deficiency; novel mutation p.Y35X; Cyt B5 gene; urine steroid profiling; methaemoglobin level; investigation; clinical management
Introduction
P450c17 is an essential enzyme in the steroidogenesis pathway. It is expressed in microsomes in steroidogenic tissues including the adrenal cortex, testis and ovary. It is the single enzyme that catalyzes both the 17 alpha-hydroxylase reaction to produce glucocorticoid, cortisol, as well as the subsequent 17,20-lyase reaction leading to production of sex steroids. While electron donation from P450- oxidoreductase (POR) is essential for proper functioning of P450c17, cytochrome B5 acts as an allosteric cofactor to facilitate electron transfer and ensure optimal 17,20-lyase function [1,2]. Isolated 17,20-lyase deficiency resulting in impaired production of testosterone is an exceedingly rare cause for 46XY Disorder of Sex Development (DSD). Only a few cases of mutations in CYP17A1 gene and cytochrome P450 oxidoreductase (POR) mutations have been identified and reported as the cause [3-7]. Kok et al [8] first reported a homozygous p.W27X mutation leading to aberrant cytochrome B5 (CytB5) protein as a cause of impaired 17,20-lyase activity in a newborn male presenting with ambiguous genitalia. The consanguineous parents carried heterozygous p.W27X mutation.
In this article, we reported the diagnosis and management of an adult woman with 46XY DSD due to a novel homozygous mutation in CYB5A inherited from her non-consanguineous parents resulting in defective testosterone production and ambiguous genitalia. We highlighted the important steps in arriving at the diagnosis and presented the surgical outcomes in the correction of external genitalia.
Case Presentation
The patient was presented to our Gynaecological Endocrinology Clinic at the age of 29 for further management of "testicular feminisation". She was noted to have ambiguous genitalia at birth with gonads being palpable at the vulva. Karyotyping revealed 46XY and gonadal biopsy confirmed the presence of testicular tissue. Diagnosis of testicular feminisation was made and she was advised to have surgical removal of gonads at puberty. However, the patient defaulted follow up since early childhood and only presented to our clinic at age 29 for clarification of the condition. She started to have breast development at 12 and axillary and pubic hair growth at 13. She had primary amenorrhea and has never attempted coitus. Physical examination revealed a body height of 170cm and weight of 48kg. Breast development was at Tanner stage IV. Axillary hair was shaved and she had a normal female pattern of pubic hair. Pelvic examination revealed clitoromegaly of 1 x 1 x 1.5 cm. A 3 x 2 cm firm mass was detected in the left labia majora and another firm mass of 2 x 2 cm was noted at the right groin, being mobile along the distal inguinal canal and right labia majora. Labia minora and vaginal mucosa were normal looking. A short blind-ended vaginal pouch of 2 cm long was detected upon gentle probing. Rectal examination revealed no palpable cervix or uterus. Ultra sonography of inguinal and groin regions confirmed the presence of testicular masses, while magnetic resonance imaging (MRI) of pelvis failed to detect any uterus or cervix.
Hormonal analysis in serum
Serum 17-alpha hydroxyprogesterone (17OHP), progesterone, testosterone, androstenedione, dehydroepiandrosterone sulphate (DHEA-S), follicle-stimulating hormone (FSH), luteinizing hormone (LH), estradiol (E2), anti-Mullerian hormone (AMH) and cortisol were measured.
Urinary steroid profiling
Urinary steroid profiling was performed by gas chromatography-mass spectrometry on a 24-hour urine collection.
Stimulation Tests
Short synacthen test and Human Chorionic Gonadotrophin (HCG) stimulation test were performed to demonstrate the extent of enzyme deficiency.
Molecular confirmation
Molecular analysis for the confirmation of the diagnosis of 17, 20-lyase deficiency was performed after obtaining written consent from the patient and her parents. DNA was extracted from peripheral whole blood using standard procedures. All the coding exons and exon-flanking introns of the CYP17A1 (NG_007955.1) and CYB5A (NG_023211.1) genes were amplified by polymerase chain reaction using the following conditions: one cycle of 94°C for 12 min; 40 cycles of denaturation at 94°C for 30 s, annealing at 63°C for 45 s, and an extension at 72°C for 45 s using the primers listed in Supplementary Table I. The reaction mixture of final volume 25 μL contained 100 ng DNA template, 1x PCR buffer (Applied Biosystems, Foster City, CA), 2.0mmol/L MgCl2, 0.2 μmol/L dNTP, 12.5 pmol of each primer, and 0.625U AmpliTaq Gold DNA polymerase. The PCR products were then purified for bidirectional DNA sequencing.
Operation
Reduction clitoroplasty and bilateral gonadectomy were performed with an aim to restore normal female external genitalia and reduce the risk of gonadoblastoma.
The operation was performed under general anaesthesia. Clitoromeglay was noted with the clitoral glans measuring 1 x 1 cm and the clitoral body measuring 3 cm long. Urethral opening was identified in its normal position. A blind-ended vaginal opening was seen below the urethral opening and was partially covered by genital fold. It admitted one finger and was ~2 cm deep. Gonads were identified at left labia majora and right groin as described above (Figure 1a).
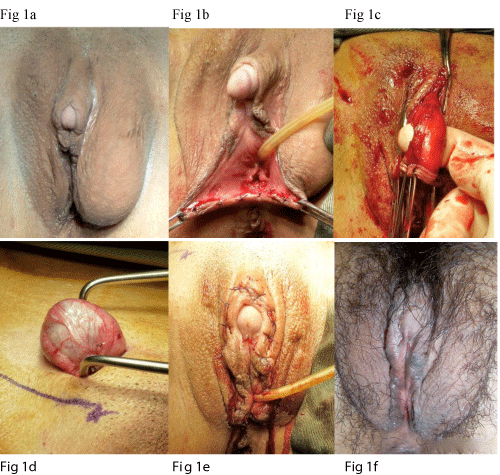
Figure 1: (a) Pre-operation - note the clitoromegaly and gonads bulging in left labial majora and right distal inguinal canal; (b) Opening of posterior fourchette to exposure the vaginal opening; (c) Corpora cavernosa was dissected free with preservation of neurovascular bundle; (d) Retrieval and excision of gonad; (e) Appearance of external genitalia at the end of operation; (f) restoration of normal female external genitalia 4 weeks after the operation.
Incision was made at midline of posterior fourchette to expose the vaginal opening (Figure 1b). Reduction clitoroplasty was performed using techniques as previously described [9]. Basically, a transverse incision was made along the clitoral body. Suspensory ligament was identified and the corpora cavernosa was dissected free with preservation of the neurovascular bundle (Figure 1c). 2cm of corpora cavernosa was removed and the cut ends were opposed and sutured.
Gonadectomy was performed through bilateral suprainguinal incision. Scarpa fascia was opened and the spermatic cords were explored and sling up (Figure 1d). Gonads were mobilized and retrieved and the gubernacular bundles were dissected.
Results
The patient recovered well after the operation and normal female external genitalia were restored (Figure 1e-f). She was put on conjugated equine estradiol 0.625 mg daily as hormonal replacement and was taught to use graduated dilators for vaginal dilatation. The vaginal canal was able to admit 1 finger easily with 3 cm deep after 6 months of dilatation.
Histology
Histological examination of the removed gonads confirmed the presence of testis, epididymis and vas deferens. There was no evidence of intratubular germ cell neoplasia, Sertoli cell adenoma or malignancy.
Hormonal Analysis and Stimulation Tests
Initial hormonal profile showed grossly elevated 17OHP, high progesterone and low testosterone, androstenedione and DHEA-S (Table 1). Serum FSH and LH levels were elevated to 128.4 and 57.5IU/L respectively. Serum E2 level was <73 pmol/L and AMH was 108.9 pmol/L. Cortisol and potassium were normal. Short synacthen test showed normal cortisol but blunted 17OHP response, while the HCG stimulation test showed blunted response in testosterone, androstenedione and DHEA-S (Table 1).
Urinary steroid profiling
Metabolites of 17-OHP, namely 17-hydroxypregnanolone and pregnanetriol, were grossly elevated while excretion of cortisol and corticosterone metabolites were within the reference intervals. All the androgen metabolites were present at very low levels. The findings suggested a blockage in the conversion of 17-hydroxypregnenolone to DHEA, and in the conversion of 17OHP to androstenedione, which is highly suggestive of isolated 17,20-lyase deficiency (Figure 2).
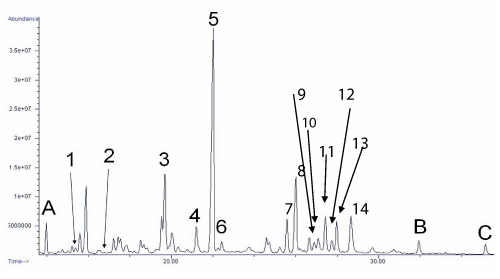
Gas chromatogram of urinary steroid profiling of the patient, showing grossly elevated metabolites of 17-hydroxyprogesterone, normal amount of cortisol and corticosterone metabolites and very small amount of androgen metabolites.
Key: A, B and C are internal standards. 1, Androsterone; 2, DHEA; 3, 17-Hydroxypregnanolone; 4, Pregnanediol; 5, Pregnanetriol; 6, Pregnenediol; 7, Pregnenetriol; 8, Tetrahydrocortisone; 9, Tetrahydrodeoxycorticosterone (THB); 10, 5 α-THB; 11, Tetrahydrocortisol (THF); 12, 5 α -THF; 13, α -cortolone; 14, β-cortolone + β -cortol
Figure 2:Gas chromatogram of urinary steroid profiling of the patient, showing grossly elevated metabolites of 17-hydroxyprogesterone, normal amount of cortisol and corticosterone metabolites and very small amount of androgen metabolites.
Key: A, B and C are internal standards. 1, Androsterone; 2, DHEA; 3, 17-Hydroxypregnanolone; 4, Pregnanediol; 5, Pregnanetriol; 6, Pregnenediol; 7, Pregnenetriol; 8, Tetrahydrocortisone; 9, Tetrahydrodeoxycorticosterone (THB); 10, 5 α-THB; 11, Tetrahydrocortisol (THF); 12, 5 α -THF; 13, α -cortolone; 14, β-cortolone + β -cortol
Methemoglobinemia
Methaemoglobin level was markedly elevated to 7.7% (Normal = 0-0.5%).
Mutational analyses
No mutation was detected in all the exons and the exon-flanking introns of the CYP17A1 gene. However, a homozygous nonsense mutation, c.105C>G was detected, changing codon 35 from TAC to TAG, i.e. p.Tyr35Term. This is a novel mutation. The results were confirmed by bidirectional sequencing. Her parents were both heterozygous for the c.105C>G, i.e. p.Y35X mutation (Figure 3).
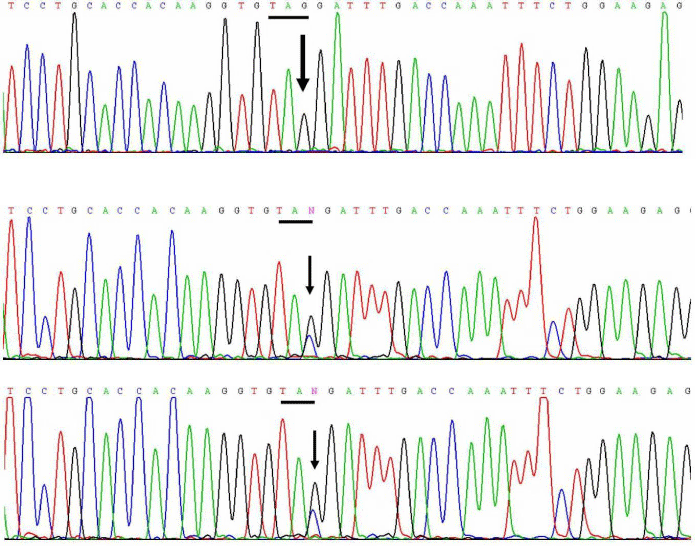
Figure 3: Electrophreograms of segment of exon 1 of the CYB5A gene of the patient (upper panel), her mother (middle panel) and her father (lower panel). The affected codon is underlined and the site of the mutation is denoted by the letter N and indicated by an arrow.
Discussion
Error(s) in testosterone biosynthesis is a rare cause for 46XY DSD and accounts for ~4% of 46XY female [10]. Testosterone is produced from its precursor, cholesterol, through a complex steroidogenesis pathway under the influence of various enzymes. Deficiency in one or more of the enzymes involved in this biosynthetic pathway could lead to clinical undermasculization of a 46XY individual, presenting with ambiguous genitalia at birth. These defects are commonly inherited as autosomal recessive traits and the phenotype ranges from partial to complete male pseudohermaphroditism.
Detailed hormonal profile including 17-OHP, DHEA-S, androstenedione and testosterone should be performed as the initial investigation. Cortisol and potassium level should also be checked to rule out cortisol or aldosterone deficiency for further investigation on the differential diagnoses. Urinary steroid profiling is a powerful test in providing definitive diagnostic information. It gives information about all adrenal steroids and the metabolites of the major hormones as well as the intermediates and precursors in a single analysis. In this case, the findings suggested a blockage in the conversion of 17-hydroxypregnenolone to DHEA, and in the conversion of 17OHP to androstenedione, which is highly suggestive of isolated 17,20-lyase deficiency. With an increasing availability of liquid chromatography tandem mass spectrometry, serum steroid profiling is also proven to be valuable for the diagnosis of various types of congenital adrenal hyperplasia [11,12]. It has been well established that both 17α-hydroxylase and 17,20-lyase activities are catalyzed by a single enzyme, P450c17. However, they are independently regulated by the differential molar ratio of P450 oxidoreductase (POR) present in adrenals and gonads, as well as the presence of Cyt B5 which serves as an alternative source of electrons that is specific for the 17,20-lyase reaction in gonads but not the 17α-hydroxylase reaction [13]. Auchus et al have also shown that CytB5 enhances 17,20-lyase activity of P450c17 without influencing 17α-hydroxylase activity, probably by affecting the activity of POR as an allosteric factor. Absence or aberrant action of CytB5 would lead to reduce but not absent 17, 20-lyase activity [2]. Most reported cases of isolated 17, 20-lyase deficiency resulted from mutations in the CYP17A1 gene and cytochrome P450 oxidoreductase (POR). A few studies also revealed a significant role of CytB5 in steroid hormone biosynthesis. An earlier study of a patient with congenital methaemoglobinaemia and DSD has revealed a splicing mutation in CytB5, leading to a 16-bp deletion in the mRNA [7]. Recently, Kok et al also reported a novel CYB5A mutation p.W27X accounting for the isolated 17, 20-lyase deficiency [8].
Mutational analysis in our patient revealed a novel homozygous nonsense mutation p. Y35X in the CYB5A gene. It echoed the findings of Kok et al [8] that CYB5A mutation is one of the responsible causes for isolated 17,20-lyase deficiency. The most curious thing about this case is that, although the parents are non-consanguineous and claimed they have different ancestral origins in the Guangdong province, they harbour the same mutation. She has four younger sisters and one younger brother, none of them has problem with sexual development. It would be interesting to perform linkage analysis on this family to demonstrate how closely the parents are related genetically. Population genetic study will help elucidate the prevalence of CYB5A mutation in our population.
Methaemoglobinaemia is also a prominent feature for those with mutation in CYB5A gene. CytB5 does not only act as an allosteric binding site for the P450c17-POR complex which is required for 17, 20-lyase activity, it is also responsible for the reduction of methaemoglobin, the oxygen-bound haemoglobin. Methaemoglobin has an increased affinity for oxygen, resulting in a reduced ability to release oxygen to tissues. With aberrant function of CytB5, reduction of methaemoglobin would be impaired and thus accumulated. Healthy people may be relatively asymptomatic with methemoglobin levels <15%. That may explain why our patient was asymptomatic for this condition all along even with a methaemoglobin level of 7.7%. However, patients with co-morbidities such as anemia, cardiovascular disease, lung disease, sepsis, or presence of other abnormal hemoglobin species (e.g. carboxyhemoglobin, sulfehemoglobin or sickle hemoglobin) may experience moderate to severe symptoms at levels as low as 5-8%.
In patients with isolated 17, 20-lyase deficiency, methaemoglobin level should be checked for two reasons. First, it serves as a simple screening test to differentiate mutations of CYP17A1 and/or P450 oxidoreductase (POR) from CYB5A gene as the aetiology; for individuals carrying the CYB5A mutation, it also acts as a baseline indicator of the degree of hypoxia.
Gender assignment is largely based on the appearance of external genitalia resulting from different degrees of enzyme deficiency and testosterone production and should be decided early. In cases with predominantly female external genitalia as in our patient, corrective surgery including reduction clitoroplasty and vulvoplasty could be performed to improve cosmesis. Attention should be paid to avoid damaging the neurovascular bundle which may result in loss of sensation or necrosis of the clitoris. Sexual function can be restored either by vaginoplasty or graduated vaginal dilatation. Although the risk of malignant change is low for non-abdominal testes, gonadectomy is usually performed to avoid such risk as well as for cosmetic reason. Hormonal replacement should be prescribed to reduce the risk of osteoporosis.
Conclusion
This case illustrated the diagnosis and clinical management of a patient having 46XY DSD due to isolated 17,20-lyase deficiency. We reported a novel homozygous mutation in CytB5A gene. Inheritance from her heterozygous non-consanguinous parents suggests that carriers for such condition(s) may exist at an incidence higher than we have believed. Clinicians are encouraged to employ urinary steroid profiling in patients presenting with ambiguous genitalia. For patients with isolated 17, 20-lyase deficiency, methaemoglobin level can be checked both to screen for CYB5A mutation before employing more sophisticated and expensive mutational tests, as well as gauging the degree of potential methaemoglobinaemia.
References
- van den Akker EL, Koper JW, Boehmer AL, Themmen AP, Verhoef-Post M, Timmerman MA, et al. Differential inhibition of 17a-hydroxylase and 17, 20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. The Journal of Clinical Endocrinology & Metabolism. 2002; 87: 5714-5721.
- Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17, 20-lyase activity of human P450c17 without direct electron transfer. J Biol Chem. 1998; 273: 3158-3165.
- Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17, 20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003; 278: 48563-48569.
- Simsek E, Ozdemir I, Lin L, Achermann JC. Isolated 17, 20-lyase (desmolase) deficiency in a 46, XX female presenting with delayed puberty. Fertil Steril. 2005; 83: 1548-1551.
- Hershkovitz E, Parvari R, Wudy SA, Hartmann MF, Gomes LG, Loewental N, et al. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17, 20-lyase deficiency. The Journal of Clinical Endocrinology & Metabolism. 2008; 93: 3584-3588.
- Biason-Lauber A, Leiberman E, Zachmann M. A Single Amino Acid Substitution in the Putative Redox Partner-Binding Site of P450c17 as Cause of Isolated 17, 20-Lyase Deficiency 1. The Journal of Clinical Endocrinology & Metabolism. 1997; 82: 3807-3812.
- Giordano SJ, Kaftory A, Steggles AW. A splicing mutation in the cytochrome b5 gene from a patient with congenital methemoglobinemia and pseudohermaphrodism. Hum Genet. 1994; 93: 568-570.
- Kok RC, Timmerman MA, Wolffenbuttel KP, Drop SL, de Jong FH. Isolated 17, 20-lyase deficiency due to the cytochrome b5 mutation W27X. J Clin Endocrinol Metab. 2010; 95: 994-999.
- Oyama IA, Steinberg AC, Holzberg AS, Maccarone JL. Reduction clitoroplasty: a technique for debulking the enlarged clitoris. J Pediatr Adolesc Gynecol. 2004; 17: 393-395.
- L Speroff FM. Clinical Gynecologic Endocrinology and infertility Lippincott Williams & Wilkins. 2005(Seventh Edition).
- Rossi C, Calton L, Hammond G, Brown HA, Wallace AM, Sacchetta P, et al. Serum steroid profiling for congenital adrenal hyperplasia using liquid chromatography-tandem mass spectrometry. Clinica Chimica Acta. 2010; 411: 222-228.
- Soldin SJ, Soldin OP. Steroid hormone analysis by tandem mass spectrometry. Clin Chem. 2009; 55: 1061-1066.
- Miller WL, Auchus RJ, Geller DH. The regulation of 17, 20 lyase activity. Steroids. 1997; 62: 133-142.