
Editorial
Austin J Pharmacol Ther. 2013;1(1): 1011.
Allosteric Modulation of G Protein-Coupled Receptors: An Emerging Approach of Drug Discovery
Christopher Wild, Kathryn A. Cunningham and Jia Zhou*
Department of Pharmacology and Toxicology, Center for Addiction Research, University of Texas Medical Branch, USA
*Corresponding author: : Jia Zhou, Department of Pharmacology and Toxicology, Center for Addiction Research, University of Texas Medical Branch, Galveston, TX 77555; USA
Received: February 03, 2014; Accepted: February 10, 2014; Published: February 27, 2014
Abstract
Allosteric modulation of G protein–coupled receptors (GPCRs) confers several significant advantages over the traditional targeting of orthosteric sites. While the field of allosteric modulation of GPCRs as we now know it will benefit from continued investigation, the explosion of interest has led to a more in–depth understanding as to precisely how allosteric modulators may usher in a new paradigm for drug discovery.
The widening gap between the cost of developing new medicines and successfully introducing therapeutics into the clinic cannot be ignored. The average R&D investment associated with bringing a drug development project to fruition is $1.2 billion over a 10–15 year time span [1]. While the annual amount spent on this endeavor has exploded since the 1950s [2], the number of approved drugs per billion U.S. dollars has decreased approximately 50–fold over the same time period [3]. Most certainly this trend cannot be attributed to a lack of technological advances considering the implementation of the high throughput capabilities in screening, sequencing and X–ray diffraction in addition to combinatorial and computational chemistry techniques available to the drug discovery team that collectively were once thought to usher in a new era in drug discovery. While the causes hampering productivity remain controversial [2–5], and the need for new targets is evident, a new paradigm with respect to how systems are targeted by small molecules is an intriguing idea that has recently gained momentum in the form of allosteric modulation. More precisely, the targeting of allosteric sites of GPCRs toward the generation of new therapeutics as a means to close the unsustainable gap between R&D costs and delivering relevant small molecules to the clinic is an exciting prospect.
The GPCRs are seven–transmembrane spanning receptors coupled to trimeric G proteins and, as a class, represent therapeutic targets for most approved medications marketed across the world yet only a small fraction of known GPCRs have been exploited for the treatment of diseases [6–8]. Traditionally, receptors of this type have been targeted with agonists or antagonists that bind to the orthosteric site that usually accommodates the endogenous ligand(s) for a given receptor. Targeting in this way can become problematic especially when the receptor belongs to a family of subtypes that share high sequence homology at the orthosteric site, usually a requisite to bind the endogenous ligand across the family of receptors. Notable examples include the metabotropic glutamate receptors (mGluRs) and serotonin receptors (5–HTRs) which are comprised of 8 and 14 receptor subtypes respectively, underscoring the importance for subtype selectivity [9,10]. This major limitation can potentially be overcome through the targeting of allosteric sites that are topologically distinct from the orthosteric sites. Conventional wisdom suggests that allosteric sites are less conserved across related receptors due to decreased evolutionary pressure that would otherwise be requisite to maintain an orthosteric binding pocket capable of accommodating the endogenous ligand(s) [11]. In short, allosteric modulation provides an opportunity to specifically target receptors that belong to a subfamily of similar GPCRs, thereby minimizing off–target effects, a significant advantage over typical agonism⁄antagonism acting at the endogenous ligand binding pocket. Moreover, most GPCR therapies are based on chronic exposure of the receptor to orthosteric ligands which raises the important issue of understanding and investigating the longterm regulatory processes of the receptors and the implications as related to decreased clinical efficacy due to desensitization and up⁄ down regulation of the target receptor. Allosteric modulators that lack intrinsic activity and only modulate the action of the orthosteric ligand may overcome the aforementioned issues, another major advantage over traditional approaches to receptor targeting. Another potential benefit of allosteric modulators is based upon the possibilitythat structural modifications can be designed to result in separate control of affinity and efficacy leading to the fine–tuning of GPCR activity in a manner that depends on the presence of the endogenous ligand [12].
The small molecule allosteric modulation of GPCRs can promote a conformational change in the receptor that often alone produces no noticeable downstream effects, but in the presence of an orthosteric ligand there can be several possible outcomes [Figure 1]: (i) positive allosteric modulators (PAMs) increase the binding affinity and⁄ or efficacy of orthosteric ligands, (ii) negative allosteric modulators (NAMs) are the antithesis of PAMs, decreasing binding affinity and⁄ or efficacy of the orthosteric ligand, (iii) silent allosteric modulators (SAMs) bind to the allosteric site without actuating a change in orthosteric binding or efficacy. The fact that allosteric modulators can lack intrinsic activity in the absence of the orthosteric ligand confers two important benefits: (i) preservation of temporal and spatial endogenous tone while fine–tuning the desired biological signaling outcome, and (ii) a ceiling effect that can minimize side effects [11]. Some allosteric modulators such as ago–PAMs can have intrinsic activity which may provide an opportunity to activate or diminish GPCR–mediated signaling in the absence of ligands that act at the orthosteric site of action. The leveraging of biased signaling (promotion of one signaling pathway at the expense of another at the same receptor) and probe dependence (differing signaling outcomes based on the identity of the chosen orthosteric ligand at a given receptor) may be exploited as novel modalities toward the treatment of disease. Yet another advantage is that so–called “undruggable” GPCRs that areactuated by intractable stimuli (i.e., large peptides) can be modulated allosterically by synthetically accessible small molecules, opening a new avenue toward targets previously unassailable [13].
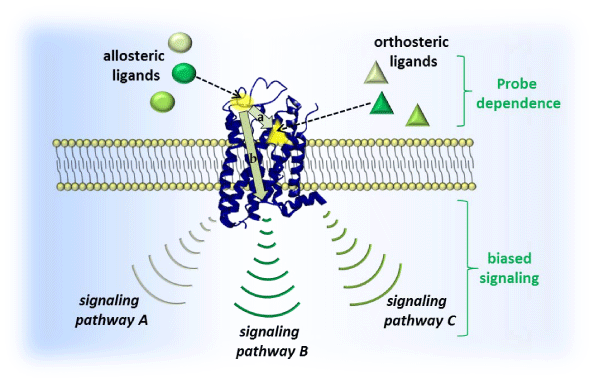
Figure 1: Schematic of GPCR signal transduction: allosteric ligands (spheres; shading represents structural diversity of different allosteric modulators that can act at same site) bind to a site (yellow sphere) that is distinctly different than the orthosteric site (yellow triangle) which accommodates an orthosteric ligand (triangles; shading represents structural diversity of different ligands that can act at the same site). Actual binding site locations are generally dictated by GPCR family type. Allosteric modulators can modulate binding affinity (a) and⁄or efficacy (b) of orthosteric ligands in a positive (PAM) or negative manner (NAM), or simply occupy the site (SAM). This modulation is often affected by the specific orthosteric ligand present (probe dependence) and can potentially alter receptor activation of certain signaling pathways at the expense of others (biased signaling).
The case for an allosteric approach for the targeting of GPCRs is building. A literature analysis of GPCR allosteric modulation provides a glimpse into the recent flurry of research in this area. Within the last decade, a steep upward trend in publications is evident (Figure 2) and will ideally translate to future therapies reaching patients. While an increase in recent interest is evident, only two allosteric modulators of GPCRs have been introduced in the clinic (Figure 3). Cinacalcet (Sensipar, Amgen, FDA–approved 2004) is a PAM that acts as a calcimimetic at the calcium–sensing receptor as a treatment for hyperparathyroidism that was designed using a homology modeling of the GPCR [14,15]. Maraviroc (Selzentry, Pfizer, FDA–approved 2007), a NAM of the chemokine receptor 5 (CCR5), is used to treat patients infected with HIV and was the result of a high throughput screening⁄medicinal chemistry program [16]. Recently the co–crystal of Maraviroc and CCR5 was solved, generating much excitement due to the disclosure of high resolution details with regard to Maraviroc binding and the resulting conformation shifts in the receptor along with the purported impact related to the HIV cell–entry mechanism [17]. While generating high resolution crystal structures of GPCRs remains a challenge, significant structural biology efforts led by Drs. R. Stevens and P. Kuhn at the Scripps Research Institute have afforded the scientific community with solved GPCR structures that provide key insights into structure–function relationships, ligand binding considerations, and the framework for molecular docking efforts as well as templates for homology modeling (www.gpcr.scripps.edu). On the pharmacology and medicinal chemistry fronts, gaps in knowledge with regard to allosteric modulators and GPCRs have been filled by efforts led by Drs. J. Conn, C. Lindsley and collaborators at the Vanderbilt Center for Neuroscience Drug Discovery with a major focus on mGluR and muscarinic receptor allosteric modulators [18]. In addition to impactful work on these specific receptors, their collaborative efforts have provided key insights on how to approach the development of GPCR allosteric modulators from the pharmacologist’s and medicinal chemist’s perspectives in general, laying the groundwork for the exploration of allosteric modulation at other GPCRs, including our own efforts to develop allosteric modulators for the serotonin 5–HT2C receptor [19].
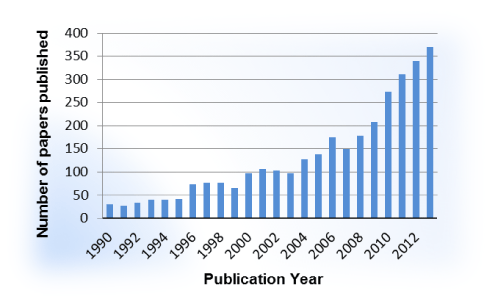
Figure 2: Number of papers published between 1990 and 2013 according to recent SciFinder search using phrase “allosteric receptor modulators”
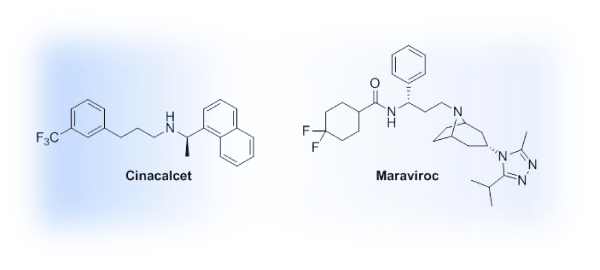
Figure 3: Structures of FDA–approved GPCR allosteric modulators.
While allosteric modulators provide new opportunities in drug design, the very advantages that have propelled this strategy into the mainstream present a set of challenges as described in the comprehensive review by an additional lead group of scientists pushing the field forward and directed by Drs. A. Christopoulos and P. Sexton at the Monash Institute of Pharmaceutical Science [13]. For instance, it is conceivable that less–conserved allosteric sites across a sub–family of receptors due to decreased evolutionary pressure at these sites, while advantageous for selectively targeting a receptor sub type, can lead to differences in the allosteric site of the same receptor between species. The potential of probe dependence of allosteric modulators necessitates careful selection of orthosteric ligands for assays. Allosteric modulator design may suffer from flat structure–activity relationships if only binding affinity and efficacy are considered without full appreciation of other key parameters such as the impact of cooperativity between allosteric and orthosteric ligand’s binding affinity and efficacy.
Despite the challenges in developing allosteric modulators, a growing body of research has emerged illuminating the advantages and methodological considerations with respect to targeting GPCRs by allosteric modulation. To date, in addition to the two marketed GPCR allosteric modulators, there are allosteric modulator compounds in clinical trials including Johnson & Johnson’s JNJ–40411813 (mGluR2 PAM, phase I) and Addex Pharma’s ADX10059 (mGluR5 NAM, phase II), demonstrating the potential of targeting the allosteric sites (www. clinicaltrials.gov). As nuanced strategies regarding pharmacological characterization and optimization of allosteric modulators continue to benefit from aggressive efforts of those in the field, future research will clarify the extent to which allosteric modulation of GPCRs – and other targets –represents a true paradigm shift in drug development. Nevertheless, allosteric modulation as a means to improve lives presents a promising approach that will ideally lead to new drugs in the years to come.
Acknowledgment
This work was supported by grants from the National Institutes of Health [P30 DA028821 (KAC), K05 DA020087 (KAC), R21 MH093844 (JZ⁄KAC)], R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, a training fellowship (T32 GM089657) awarded to the Keck Center for Interdisciplinary Bioscience Training of the GCC (CW), John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at the University of Texas Medical Branch (UTMB).
References
- https://www.phrma.org/industryproile2013. Accessed: 2013 November 01.
- Munos B. Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov. 2009; 8: 959-968.
- Scannell J W, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D eficiency. Nat Rev Drug Discov. 2012; 11: 191-200.
- Dickson M, Gagnon JP. Key factors in the rising cost of new drug discovery and development. Nat Rev Drug Discov. 2004; 3: 417-429.
- Southan C, Varkonyi P, Boppana K, Jagarlapudi SA, Muresan S. Tracking 20 Years of Compound-to-Target Output from Literature and Patents. PLoS One. 2013; 8: e77142.
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein- coupled receptor target family. ChemMedChem. 2006; 1: 761-782.
- Conn PJ.; Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009; 8: 41-54.
- Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis. 2013; doi: 10.1016/j. nbd.2013.09.013.
- Menniti FS, Lindsley CW, Conn PJ, Pandit J, Zagouras P, et al., Allosteric Modulators for the Treatment of Schizophrenia:Targeting Glutamatergic Networks. Curr Top Med Chem. 2013; 13: 26-54.
- Bubar M, Cunningham K. Prospects for serotonin 5-HT2R pharmacotherapy in psychostimulant abuse. Progress in Brain Research. 2008; 172: 319-346.
- Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, et al., Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012; 55: 1445-1464.
- Kenakin TP. ‘7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009; 30: 460-469.
- Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013; 12: 630-644.
- Nemeth EF. Allosteric modulators of the extracellular calcium receptor. Drug Discov Today Technol. 2013; 10: e277-284.
- Harrington PE, Fotsch C. Calcium Sensing Receptor Activators: Calcimimetics. Curr Med Chem. 2007; 14: 3027-3034.
- Dorr P, Westby M, Dobbs S, Grifin P, Irvine B, et al., Maraviroc (UK- 427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeiciency virus type 1 activity. Antimicrob Agents Chemother. 2005; 49: 4721-4732.
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, et al., Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013; 341: 1387- 1390.
- Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008; 3: 530-541.
- Ding C, Bremer NM, Smith TD, Seitz PK, Anastasio NC, et al., Exploration of Synthetic Approaches and Pharmacological Evaluation of PNU-69176E and its Stereoisomer as 5-HT Receptor Allosteric Modulators. ACS Chem Neurosci. 2012; 3: 538-545.