
Research Article
Austin J Proteomics Bioinform & Genomics. 2023; 8(1): 1034.
A Metagenomics Insight in the Cyanosphere of Edible Andean Macrocolonies (Llayta)
Claudia Vilo C1,2; Galetovic A1; Dong Q3; Gómez-Silva B1*
¹Laboratory of Biochemistry, Biomedical Department, Health Sciences Faculty and Centre for Biotechnology and Bioengineering (CeBiB), Universidad de Antofagasta, Antofagasta, Chile
²Laboratory of Molecular and Cellular Biology of Cancer, Department of Biomedical Sciences, Faculty of Medicine, Universidad Católica del Norte, Coquimbo, Chile
³Center for Biomedical Informatics, Department of Medicine, Stritch School of Medicine, Loyola University of Chicago, Maywood, Illinois, USA
*Corresponding author: Gómez-Silva B Biomedical Department, Health Sciences Faculty and Centre for Biotechnology and Bioengineering (CeBiB), Universidad de Antofagasta, Antofagasta, Chile. Email: [email protected]
Received: July 26, 2023 Accepted: August 25, 2023 Published: September 01, 2023
Abstract
It is well stablished that some cyanobacterial biomasses have nutritious ingredients and have been consumed for centuries. In South America, macrocolonies of filamentous diazotrophic species, order Nostocales, known as Llayta, can be found at Andean wetlands and have been consumed since pre-Columbian times. Cyanocohniella sp. LLY has been identified as a major cyanobacterial member of the Llayta cyanosphere, however, the microflora colonizing these macrocolonies has been poorly explored. Genomic DNA from Gentamycin-treated cyanobacterial filaments purified from the rehydrated biomass of Llayta macrocolonies, were subjected to metagenomics studies to identify resilient members of the accompanying heterotrophic bacterial flora. Here, we report a metagenomics-based identification of five prominent bacterial members belonging to the genera Mesorhizobium, Microvirga, Paracoccus, Aquimonas, and Blastomonas, tightly adhered to Llayta trichomes. Their metagenome-assembled genomes and information on putative genes and genes clusters involved in primary and secondary metabolism is also provided. We expect this information on Llayta cyanosphere would help to further explore adaptive responses and role of cyanobacteria macrocolonies in ecosystem processes under the stressful environmental conditions prevailing at the Atacama Desert highlands.
Keywords: Andes wetlands; Cyanosphere; Edible cyanobacterial macrocolonies; Llayta; Metagenomics; Metal resistance genes; Microbiota; Nostocales
Introduction
Consumption of edible macrocolonies of filamentous cyanobacteria, locally known as Llayta, is a centenary Andean alimentary practice in South America [1-3]. Llayta macrocolonies can be found at Andean wetlands above 3,000 m of altitude, at sites not always readily accessible [4,5], but it is available nowadays as a dried natural product at food markets in northern Chile and southern Peru [6]. Also, Llayta is considered a safe natural food ingredient based on its biochemical content, absence of cyanotoxins, ethnographic testimonies, and absence of negative epidemiological evidence [4-7].
Filamentous diazotrophic cyanobacteria are ubiquitous to almost any ecosystem on Earth, including some under severe environmental conditions. Trichomes containing vegetative cells and N2-fixing heterocyst develop during the life cycle of diazotrophic Nostoc species and can form green to brown sheet-like or spherical macrocolony, depending up-on the species, dehydration state, and location [8,9]. In natural environments, colonial and filamentous cyanobacteria can be colonized by microorganisms emplaced around, within or outside the host colony, with varied composition [10-14], and involved in biogeochemical cycles, ecosystem services, and algicidal or stimulatory effects [10,14-16].
Recently, Vilo et al. [16] reported an improved metagenomics analysis identifying Cyanocohniella sp. LLY as the potential cyanobacterium responsible for forming Llayta macrocolonies at the Andes wetlands, providing the first Metagenome-Assembled Genome (MAG) for this genus. Nonetheless, Llayta cyanosphere is still a pending issue. The identi-fication and draft genome of a Bacillus bacterium from the microbiota associated to Llayta colonies [17] was the first approach to address physiological relationships within the Llayta microbiota and to gain insights into the survival and adaptive strategies to dryness, metals, metalloids, and UV radiation, among other prevalent extreme environmental con-ditions at the Andes wetlands.
Metagenomics is proper culture-independent tool that allows insights into the composition and genomic capabilities of the microbial community from natural settings [18-20], and prospection of natural products [18-21]. To improve our under standing on Llayta cyanosphere, we focused our metagenomics-based study on the resilient bacterial microflora associated with isolated, Gentamycin-treated Llayta filaments. Here we present the identification of microbial taxa tightly adhered to Llayta trichomes, the genome recon-struction of five prominent bacterial members, the identification and annotation of func-tional genes, and an insight into their metabolic capabilities.
Materials and Methods
Isolation of Llayta Filaments, Growth Conditions and Antibiotic Treatment
Dry biomass of Llayta macrocolonies were obtained during 2015 at the major farmer´s food market in Arica, Chile. Filament isolation procedures have been previously described [17]. Briefly, rehydrated Llayta samples were grown and enriched in liquid, ni-trogen-free Arnon mineral medium [4,5,22]. Aliquots were seeded in agar plates, isolated filaments were retrieved under a stereo microscope, grown in fresh medium as above, harvested after 3 weeks at the exponential growth phase, washed with fresh medium, and recovered as a pellet by centrifugation at 4,000 x g for 5 min, at room temperature. Our metagenomics studies were focused on genomic DNA recovered from bacteria closely associated to isolated Llayta trichomes. Then, metagenomics analyses were conducted on trichomes previously incubated with Gentamycin in the presence of carbon sources to stimulate growth of heterotrophic bacteria and in darkness to slow down cyanobacterial metabolic activity. Approximately, 10-15 mL of cultured Llayta filaments were collected at the exponential growth phase, washed with fresh medium, and suspended in 20 mL of fresh Arnon medium containing Gentamycin (1 mg/mL; Sigma Aldrich, Chile), Casamino acids (1.6 mg/mL; Sigma Aldrich, Chile), and D-glucose (0.8 mg/mL; Sigma Aldrich, Chile), and incubated for 48h in darkness at 30°C, and gently stirred (120 rpm). After this treatment, microbial contamination of Llayta filaments decreased by nearly four orders of magnitude (Gentamycin was the most efficient antibiotic tested). The biomass was recovered by centrifugation and used to extract total genomic DNA.
DNA Extraction and Sequencing
Total genomic DNA from the filament pellets was extracted with Ultra Clean Micro-bial DNA isolation kit (MoBio Labs. Inc., Carlsbad, CA, USA), following the manufactur-er´s instructions. DNA quality was evaluated by electrophoresis in 0.8 % agarose gel and quantified photometrically at 260 nm. The Llayta metagenome was sequenced via MiSeq sequencing technology using shotgun paired-end libraries, with an average insert size of 250 bp. Reads had an average length of 300 bp, with good quality scores, as evaluated by the FastQC program (version 0.10.0). The sequencing produced a total of 17,137,246 reads. Sequencing reads are available at the Sequence Read Archive (SRA) with accession num-ber SRR17916224. The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAKOMP000000000, JAKOMQ000000000, JAKOMR000000000, JAKOMS000000000, JAKOMT000000000, and JAKOMU000000000.
Bioinformatic Analysis
The metagenomic sequences were submitted to the Rapid Annotation using Subsys-tems Technology for Metagenomes (MG-RAST) web server [23] for a taxonomic and func-tional assignment using default parameters. In addition, metagenomic assembly was done using MEGAHIT assembler v.1.2.9 [24], and binning was conducted using the PATRIC web server [25]. The complete genome was annotated using the Rapid Annotations using Subsystem Technology (RAST) server version 4.0. Secondary metabolites were searched with PRISM version 4.4.5 [26] and AntiSMASH version 6.1.1 software [27]. In addition, in-house BLAST analysis was done against customized metal resistance genes databases.
Results and Discussion
During our work, an abundant microflora with high bacterial titers was detected in attempts to isolate and purify axenic Llayta filaments; however, incubation of isolated Llayta filaments with Gentamycin decreased such titers by nearly four orders of magnitude. Then, we focused our metagenomics-based study on the resilient bacterial microflora associated with isolated and Gentamycin-treated Llayta filaments to improve our under-standing on Llayta cyanosphere.
Genomic DNA from Gentamycin-treated cyanobacterial filaments, purified from the rehydrated biomass of Llayta macrocolonies, were subjected to metagenomics studies to identify resilient members of the accompanying heterotrophic bacterial flora, construct the corresponding metagenomic-assembled genomes, and gain insights into their functional metabolic capabilities. We identified the antibiotic-resilient bacterial community associ-ated to Llayta filaments, reconstructed the genomes of five prominent bacteria, and de-tected putative biosynthetic genes related to primary and secondary metabolism and adaptive stress strategies.
Microbial Diversity in Llayta Trichomes
Taxonomic assignments obtained from metagenomics analyses of shotgun meta-genome showed that the microbiota associated with Llayta filaments was dominated by bacteria (99%), while representatives of the domains Archaea (0.2%) and Eukarya (0.1%) were present at a much lower extent. Predominant bacteria in Llayta trichomes belong to the phyla Proteobacteria (82%) and Cyanobacteria (16%) (Figure 1). Dominant genera were Xanthomonas (38%), Stenotrophomonas (15%), Methylobacterium (9.3%), Nostoc (40%), Anabaena (26%), and Nodularia (21%). Thus, Xanthomonas (38%) and Nostoc (40%) were the primary genera (Figure 1).
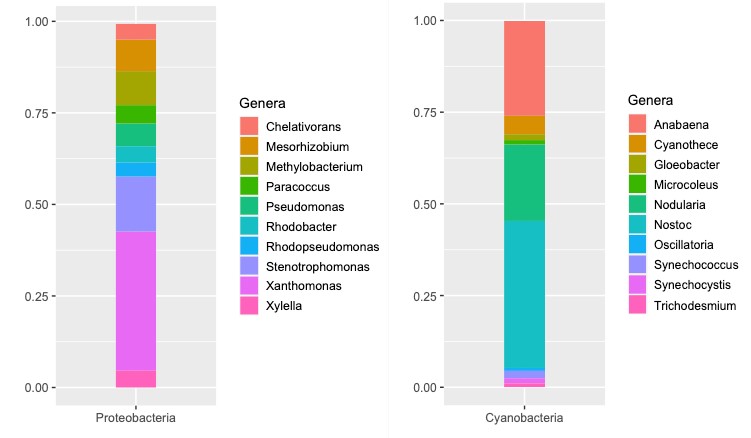
Figure 1: Genus-level relative abundance of the ten dominant Proteobacteria and Cya-nobacteria found in the Gentamycin-treated Llayta filaments.
Five bacteria associated with isolated Llayta filaments were identified by metagenomics analysis: two nitrogen-fixing Alphaproteobacteria (genera Mesorhizobium and Microvirga), one aerobic photoheterotrophic Alphaproteobacteria (genus Blastomonas), one Gammaproteobacteria (genus Aquimonas) and one nitrogen denitrifying bacterium (genus Paracoccus). The summary statistics for the assembled MAGs and the corresponding phy-logenetic analyses by the Maximum Likelihood method based on 16S rRNA gene se-quences are shown in Table 1 and Figure 2, respectively.
![]()
Genus
Sequence
sizeContigs number
N50
valueCompleteness (%)
Contamination
(%)Aquimonas (gamma-proteobacterium)
4,464,829
80
155,856
98.3
1.4
Blastomonas (aerobic photoheterotrophic alphaproteobacteria)
3,312,218
641
7,513
98.1
15.2
Mesorhizobium (N2-fixing alpha-proteobacterium)
4,745,523
165
427,465
91.3
11.1
Microvirga (N2-fixing alpha-proteobacterium)
3,719,434
70
273,976
98.4
2.8
Paracoccus (nitrogen denitrifying bacterium)
4,116,456
107
139,894
100
0.0
Table 1: Statistics for assembling and binning of the reconstructed metagenomes of prominent bacteria members associated with Llayta filaments.
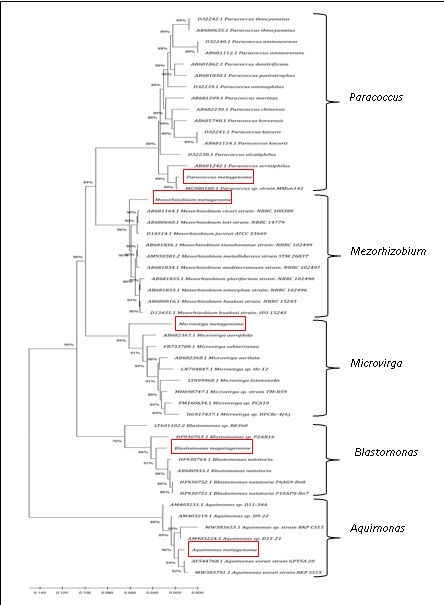
Figure 2: Phylogenetic tree of 16S rRNA gene depicting clades of the five assembled bacteria genera, Aquimonas, Blastomonas, Microvirga, Mesorhizobium, and Paracoccus.
Paracoccus was the only bacterial genus associated with Llayta filaments that coincides with Paracoccus strain B50 found as a culturable bacterial isolate associated to Nodularia spumigena filaments, which negatively affected the cyanobacterium growth [14]. We also identified a member of the genus Anabaena on the resilient microflora accompanying the Llayta cyanosphere, in agreement with previous observations reported on Nostoc colonies from Parinacota wetlands [12]. Recent studies have reported on the diversity and em-placement of heterotrophic bacteria in Nostoc macrocolonies. Secker et al. [11] showed ab-sence of bacteria at the inner matrix of Nostoc macrocolonies from ephemeral wetlands in New Zealand, while high bacterial diversity was found at their external surface, enriched with members of the genus Sphingomonas. Conversely, a different bacterial composition was found at the inner matrix, outer layer, and the littoral zone of Nostoc macrocolonies from Chungará Lake in northern Chile [12]. Comparatively, and using metagenomics, multiple taxonomic markers, and microscopic approaches, Satjarak et al. [13] reported high taxonomic diversity (cyanobacteria, microalgae, and anoxygenic bacterial genera) on the accompanying epimicrobiota of macroscopic dark-brown sheets of Nostoc from standing water pools at Parinacota, Lauca National Park, in northern Chile.
Metal Resistance Capabilities of the Llayta MAGs
The reconstructed Llayta MAGs were analyzed with the RAST server for genome an-notation based on subsystem annotations. A total of 156 putative metal resistance genes was also observed in the Llayta microbiota (Table 2) indicating the putative adaptability of microbial communities to the volcanic-rich activity of the Andes Mountains. We observed genes for copper homeostasis and tolerance to mercury and chromium compounds (Table 2). All MAGs analyzed showed the presence of components of the cop operon associated with cytoplasmic copper levels control [28]. Blastomonas MAG contains genes coding the copper resistance protein B, which showed a 79% identity with Blastomonas sp. protein (accession number WAC22814.1). Copper resistance protein B has been described to promote copper sequestrating at the outer membrane. Microvirga and Paracoccus MAGs had genes coding for protein CopG associated with copper binding [28]. Microvirga MAG showed a 100% identity with conserved domain DUF411 from Microvirga sp. protein (accession number MCG6123028.1), which has been classified as a CopG-like protein [29]. Paracoccus MAG showed three copies of CopG gene, with 100% identity to Paracoccus sp. DUF411 domain-containing proteins, and metal binding proteins (accession numbers: MCG6113278.1, MCG6112398.1, and MCG6112409.1). Genes coding for proteins of the cut family involved in copper homeostasis (uptake, storage, delivery, and efflux) [28] were observed. MAGs from Blastomonas, Microvirga, Mesorhizobium, and Paracoccus contained genes coding for CutE protein related to copper storage and transport [30] (100% identity with Blastomonas sp., accession number MCG6119876.1; 100% identity with Microvirga sp., accession number MCG6121847.1; 100% identity with Mesorhizobium sp., accession number MCG6114269.1; and 100% identity with Paracoccus sp., accession number MCG6110613.1, respectively). All MAGs showed the presence of czc operon associated with cobalt-zinc-cadmium resistance and export pathways for Cd2+, Zn2+, and Co2+ ions [31]. Finally, all MAGs had putative capabilities for coding CzcD protein involved in reg-ulating the Czc system and CzcA protein, a metal cation efflux transporter [30,31]. Regarding arsenic resistance, arsC gene and arsenical-resistance protein ACR3 were present all MAGs. The arsC gene translates into arsenate reductase enzyme, which catalyzes the reduction of arsenate to arsenite to be extruded from the cell by arsenite permeases such as ACR3 [32,33].
![]()
Metal resistance
Blastomonas
Aquimonas
Mesorhizobium
Microvirga
Paracoccus
Copper homeostasis and tolerance
10
5
13
12
11
Cobalt-zinc-cadmium resistance
6
4
6
7
8
Mercuric
resistance4
2
4
4
4
Resistance to chromium compounds
1
1
1
2
1
Arsenic resistance and detoxification
8
5
16
13
8
Table 2: Number of genes associated with metal resistance in Llayta MAGs.
Secondary Metabolism of the Llayta MAGs
Secondary metabolism analysis of the five reconstructed MAGs showed specific adaptations to overcome some of the environmental conditions found at the Atacama Desert highlands. Paracoccus MAG contained the gene sequence associated with ectoine synthase [34], showing 100% identity to ectoine synthase gene from Paracoccus hibiscisoli (accession number WP_136857538). Blastomonas MAG showed genes coding for lasso peptides B1 and B2. MAGs from Aquimonas, Mesorhizobium, and Paracoccus showed the presence of genes coding for phytoene synthase and lycopene cyclase.
On putative carotenoid biosynthetic capabilities [35], lycopene cyclase and phytoene synthase coding genes in Aquimonas MAG showed 100% identity to lycopene cyclase fam-ily protein and phytoene synthase family protein from Aquimonas sp. (accession numbers MCG6118585 and MCG6118584, respectively). Phytoene synthase gene sequence from Mesorhizobium MAG showed 100% identity to phytoene/squalene synthase family protein from Mesorhizobium sp. (accession number MCG6115296). In Paracoccus MAG, the phy-toene synthase and lycopene cyclase coding gene showed 100% sequence identity to phy-toene synthase protein, and lycopene beta cyclase CrtY reported for Paracoccus sp. (acces-sion numbers MCG6110761 and MCG6110759, respectively).
The inferred functional capabilities of the Llayta microbiota from their metagenomic content may be important to improve our understanding on the adaptive responses of this microbial community to survive and proliferate under the stressful environmental conditions found in the Andean wetlands. For instance, we found evidence of ectoine production on Paracoccus MAG, indicating specific adaptations to high salinity and dessication. Ectoine is a known natural compound that has been associated to osmoregulation in bacteria [34]. Also, carotenoids biosynthesis was observed in Aquimonas, Mesorhizobium, and Paracoccus MAGs, indicating the need for protection against UV radiation, a well-known hazard in the Atacama Desert, particularly on the Andean plateau [36,37]. In addition, metalloid resistance seems to be a common trait among Atacama Desert microorganisms, which may be critical for those microbial communities to acquire arsenic reduction and extrusion, copper sequestration, and cobalt, zinc, and cadmium outer extrusion.
Conclusions
Members of the gentamycin-resilient microbial community associated with cyanobacterial macrocolonies of Llayta filaments were identified using a culture-independent approach. Metagenomics analyses allowed the reconstruction and draft genomes for five prominent bacteria. This information provides an insight into microbial functional capabilities, biosynthetic pathways, and adaptive strategies to the environmental conditions at high-altitude Andean wetlands. Beyond the well-known, centuries-old consumption of Llayta macrocolonies, their cyanosphere opens new opportunities for biotechnological applications. Finally, the cyanosphere associated to filaments of edible Llayta macrocolonies is another example of the microbial richness present at the Atacama Desert; such microbiome deserves extensive research to better understand its role in drylands ecosystem processes [38,39].
Author Statements
Funding Source
This research was funded by ANID, Chile, grant CeBiB FB-0001. The funding organization had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Data Archiving
The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAKOMP000000000, JAKOMQ000000000, JAKOMR000000000, JAKOMS000000000, JAKOMT000000000, and JAKOMU000000000.
Data Availability
Data will be made available on request.
Credit Authorship Contribution Statement
Benito Gómez-Silva: Conceptualization, Investigation, Writing-original draft preparation, Writing-review and editing, Project administration, Funding acquisition. Claudia Vilo: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing-original draft preparation, Writing-review and editing. Alexandra Galetovic: Methodology, Validation, Investigation, Data curation, Writing-review and editing. Qunfeng Dong: Validation, writing-review and editing. All authors have read and agreed to the published version of the manuscript.
Declaration of Compiting Interest
The authors declare no conflict of interest.
Acknowledgments
Data was generated in the Genome Sequencing Facility, which is supported by UT Health San Antonio, NIH-NCI P30 CA054174 (Cancer Center at UT Health San Antonio), NIH Shared Instrument grant 1S10OD021805-01.
References
- Bertonio L. Vocabulario de la Lengua Aymara. Colección biblioteca nacional de Chile. Perú: Impreso en la Compañía de Jesús. Electronic document; 1612. Available from: http://www.memoriachilena.cl/602/w3-article-8656.html.
- Lagerheim MG. La Yuyucha. La Nuova Notarisia. 1892; 3: 1376-7.
- Mannheim B. Poetic form in Guaman Poma’s Wariqsa Arawi. Amerindian. 1986; 11: 41-67.
- Gómez-Silva B, Mendizabal I, Tapia I, Olivares H. Microalgae del norte de Chile. IV. Composición química de Nostoc commune Llaita. Rev Investig Cient Tecnol CS. March 1994;3:19-25.
- Galetovic A, Araya JE, Gómez-Silva B. Composición bioquímica y toxicidad de colonias comestibles de la cianobacteria andina Nostoc sp. Llayta. Rev Chil Nutr. 2017; 44: 360-70.
- Rivera M, Galetovic A, Licuime R, Gómez-Silva B. A microethnographic and ethnobotanical approach to Llayta consumption among the Andes feeding practices. Foods. 2018; 7: 202.
- Galetovic A, Azevedo J, Castelo-Branco R, Oliveira F, Gómez-Silva B, Vasconcelos V. Absence of cyanotoxins in Llayta, edible Nostocaceae colonies from the Andes Highlands. Toxins. 2020; 12: 382.
- Dodds WK, Gudder DA, Mollenhauer D. The ecology of Nostoc. J Phycol. 1995; 31: 2-18.
- Sand-Jensen K. Ecophysiology of gelatinous Nostoc colonies: unprecedented slow growth and survival in resource poor and harsh environments. Ann Bot. 2014; 114: 17-33.
- Gao S, Kong Y, Yu J, Miao L, Ji L, Song L, et al. Isolation of axenic cyanobacterium and the promoting effect of associated bacterium on axenic cyanobacterium. BMC Biotechnol. 2020; 20: 61.
- Secker NH, Chua JPS, Laurie RE. Characterization of the cyanobacteria and associated bacterial community from an ephemeral wetland in New Zealand. J Phycol. 2017; 52: 761-73.
- Aguilar P, Dorador C, Vila I, Sommaruga R. Bacterial communities associated with spherical Nostoc macrocolonies. Front Microbiol. 2019; 10: 483.
- Satjarak A, Graham LE, Piotrowski MJ, Trest MT, Wilcox LW, Cook ME, et al. Shotgun metagenomics and microscopy indicate diverse cyanophytes, other bacteria, and microeukaryotes in the epimicrobiota of a northern Chilean wetland Nostoc (cyanobacteria). J Phycol. 2021; 57: 39-50.
- Salomon PS, Janson S, Granéli E. Molecular identification of bacteria associated with filaments of Nodularia spumigena and their effect on the cyanobacterial growth. Harmful Algae. 2003; 2: 261-72.
- Ducklow H. Microbial services: challenges for microbial ecologists in a changing world. Aquat Microb Ecol. 2008; 53: 13-9.
- Vilo C, Dong Q, Galetovic A, Gómez-Silva B. Metagenome-assembled genome of Cyanocohniella sp. LLY from the Cyanosphere of Llayta, an Edible Andean Cyanobacterial Macrocolony. Microorganisms. 2022;10(8):1517.
- Vilo C, Galetovic A, Araya JE, Gómez-Silva B, Dong Q. Draft genome sequence of a Bacillus bacterium from the Atacama Desert wetlands metagenome. Genome Announc. 2015; 3: e00955-15.
- Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Rev. 2004; 68: 669-85.
- Gómez-Silva B, Vilo-Muñoz C, Galetovic A, Dong Q, Castelán-Sánchez HG, Pérez-Llano Y, et al. Metagenomics of Atacama lithobiontic extremophile life unveils highlights on fungal communities, biogeochemical cycles and carbohydrate-active enzymes. Microorganisms. 2019; 7: 619.
- Thomas T, Gilbert J, Meyer F. Metagenomics – a guide from sampling to data analysis. Microb Inform Exp. 2012; 2: 3.
- Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai HC, et al. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat Chem Biol. 2017; 13: 470-8.
- Arnon DI, McSwain BD, Tsujimoto HY, Wada K. Photochemical activity and components of membrane prepa-rations from blue-green algae. I. Coexistence of two photosystems in relation to chlorophyll a, and removal of phycocyanin. Biochim Biophys Acta. 1974; 357: 231-45.
- Keegan KP, Glass EM, Meyer F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. In: Microbial environmental genomics (MEG). New York: Humana Press; 2016: 207-33.
- Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, et al. MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016; 102: 3-11.
- Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014; 42: D581-91.
- Skinnider MA, Merwin NJ, Johnston CW, Magarvey NA. PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017; 45: W49-54.
- Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, et al. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021; 49: W29-35.
- Puig S, Rees EM, Thiele DJ. The ABCDs of periplasmic copper trafficking. Structure. 2002; 10: 1292-5.
- Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017; 45: D200-3.
- Li J, Ji C, Chen J, Yang Z, Wang Y, Fei X, et al. Identification and characterization of a novel Cut family cDNA that encodes human copper transporter protein CutC. Biochem Biophys Res Commun. 2005; 337: 179-83.
- Osman GEH, Abulreesh HH, Elbanna K, Shaaban MR, Samreen S, Ahmad I. Recent progress in metal-microbe interactions: prospects in bioremediation. J Pure Appl Microbiol. 2019; 13: 13-26.
- Silver S, Phung LT. Genes and enzymes involved in bacterial oxidation and reduction of inorganic arsenic. Appl Environ Microbiol. 2005; 71: 599-608.
- Mukhopadhyay R, Rosen BP. Arsenate reductases in prokaryotes and eukaryotes. Environ Health Perspect. 2002; 110: 745-8.
- Jebbar M, Talibart R, Gloux K, Bernard T, Blanco C. Osmoprotection of Escherichia coli by ectoine: uptake and accumulation characteristics. J Bacteriol. 1992; 174: 5027-35.
- Sandmann G. Molecular evolution of carotenoid biosynthesis from bacteria to plants. Physiol Plant. 2002; 116: 431-40.
- Cordero RR, Damiani A, Jorquera J, Sepúlveda E, Caballero M, Fernandez S, et al. Ultraviolet radiation in the Atacama Desert. Antonie Leeuwenhoek. 2018; 111: 1301-13.
- Gómez-Silva B. Lithobiontic life: Atacama rocks are well and alive. Antoine Leeuwenhoek. 2018; 111: 1333-43.
- Gómez-Silva B, Batista-García RA. The Atacama Desert: A Biodiversity Hotspot and not just a mineral-rich region. Front Microbiol. 2022; 13: 812842.
- Archer SDJ, Lee KC, Caruso T, Alcami A, Araya JG, Craig Cary S, et al. Contribution of the soil bacteria to the atmosphere across biomes. Sci Tota Environ. 2023; 871: 163137.