
Department of Surgery, University of Pittsburgh Cancer Institute, USA
*Corresponding author: Rui Kang and Daolin Tang, Department of Surgery, University of Pittsburgh Cancer Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15213, USA
Received: August 08, 2014; Accepted: December 03, 2014; Published: December 03, 2014
Citation: Tang D, Lotze M, Zeh HJ and Kang R. Targeting HMGB1 in Acute Pancreatitis. Austin J Surg. 2014;1(9): 1045. ISSN: 2381-9030.
High Mobility Group Box 1 (HMGB1) is normally a non-histone nuclear protein that acts as a DNA chaperone with DNA binding and bending activity. Besides its intracellular function, extracellular HMGB1 is an inducer, sensor, mediator, and effect or in the innate immune response to infection and sterile inflammation. We recently demonstrated that HMGB1 is an important regulator of the links between local tissue injuries and the systemic inflammatory response in acute pancreatitis. Deficiency of endogenous pancreatic HMGB1 in experimental acute pancreatitis leads to oxidative stress-mediated nuclear catastrophe and nucleosome (including histone and DNA) release, which then recruits and activates macrophages, with subsequent HMGB1 release locally and into the circulation. Neutralizing extracellular histone and HMGB1 protects against acute pancreatitis in conditional pancreas-specific HMGB1 knockout mice. Thus, HMGB1 has a dual role in the pathogenesis of pancreatitis, shedding light on the role of the innate immune response in infection and tissue damage.
Keywords: HMGB1; DAMPs; Acute Pancreatitis; sRAGE
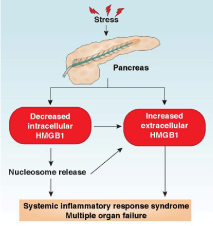
Acute pancreatitis is an inflammatory condition of the pancreas often caused by gallstones and heavy alcohol abuse. Acute pancreatitis exhibits a broad clinical spectrum of findings varying from mild and self-limited to severe, catastrophic illness. It’s most severe form, necrotizing pancreatitis, presents local pancreatic injury, multiple organ failure, a systemic inflammatory response, and high mortality rates [1]. Under normal conditions, digestive enzymes or proteases (e.g., trypsin, elastase, and lipase) are stored in zymogen granules in an inactive form. In pancreatitis, these proteases are activated within the pancreatic acinar cells and lead to organelle injury and auto digestion of the pancreas [2]. Apart from local tissue injury, Systemic Inflammatory Response Syndrome (SIRS) is the major pathobiological process responsible for the morbidity and mortality of severe acute pancreatitis. Acute pancreatitis is usually a sterile inflammatory disorder involving a complex cascade of interacting immune cells and inflammatory mediators. Several inflammatory mediators are initially released by pancreatic acinar cells and result in the recruitment and activation of neutrophils, monocytes, and macrophages [3-6], which lead to further inflammatory mediator release, acinar cell injury, and SIRS [7]. Damage-Associated Molecular Pattern molecules (DAMPs) are endogenous molecules released by dead, dying, and injured cells that mediate the response to inflammation and immunity through various DAMP receptors such as Toll-Like Receptors (TLRs) and the Receptor for Advanced Glycation End product (RAGE) [8- 10]. Increasing evidence indicates that DAMPs (including nuclear and mitochondrial DAMPs) promote the progression from local pancreatic damage to SIRS in acute pancreatitis; play a central role in the pathogenesis of multisystem organ failure; and could constitute a promising molecular target for therapeutic approaches to acute pancreatitis [11]. Our recent study implicates that High Mobility Group Box 1 (HMGB1), a classical nuclear DAMP, plays a dual role in acute pancreatitis (Figure 1). In this mini-review, we will introduce the basic function of HMGB1 and focus on how HMGB1 regulates the development of acute pancreatitis.
HMGB1 is a member of a family of highly-conserved proteins containing HMG box domains among species. It has two homologous HMG boxes (termed box A and box B) and one short acidic tail. HMGB1 has multiple functions in health and disease, depending on its location and modification [12]. In the nucleus, HMGB1 is a non-histone chromosome protein that binds to DNA, facilitating numerous nuclear events such as maintenance of genome stability, nucleosome sliding, gene transcription, DNA repair, and V(D)J recombination [13-16]. In the cytosol, HMGB1 is a positive autophagy regulator that binds to Beclin-1 protein, contributing to autophagosome initiation [17-19]. In the cell membrane, HMGB1 facilitates cell migration and neurite outgrowth in a RAGE-dependent manner. In addition to intracellular function, HMGB1 can also act as a DAMP when passively released from injured cells or actively secreted from immune cells [8,20-22]. Once released, HMGB1 binds to a number of receptors (e.g., TLR2, TLR4, TLR9, and RAGE) and mediates the recruitment of inflammatory cells and the release of proinflammatory cytokines (e.g., tumor necrosis factor-α and interleukin-6) [23]. Apart from a direct receptor interaction, HMGB1 can be taken up by cancer and immune cells, triggering energy metabolism and inflammasome activation, respectively [24,25]. Special attention has been paid in recent years to the regulation of extracellular HMGB1 activity by its partner, receptor, redox status, and cleavage [12]. For example, extracellular HMGB1 can exist in a hetero complex with other molecules, such as interleukin-1, C-X-C motif chemokine 12, DNA, nucleosome, orlipopolysaccharide, that produce synergistic effects in many cell processes [26]. Reduced HMGB1 triggers autophagy and has immune activity, whereas oxidized HMGB1 induces apoptosis and loses immune activity [27]. Overall, serum HMGB1 levels are elevated in patients with many diseases, especially inflammation-associated diseases, and may serve as a valuable biomarker [12]. More studies are needed to clearly elucidate the role of HMGB1 in disease.
Several studies have confirmed that serum levels of HMGB1 are significantly elevated and correlate with the severity of patients with acute pancreatitis [28,29]. For instance, Yasuda et al. observed that serum HMGB1 levels in patients with severe acute pancreatitis (5.4±1.3 ng/mL) on admission (within 72 hours after onset) was higher than those in healthy subjects (1.7±0.3 ng/mL) [28]. Kocsis et al. reported that circulating HMGB1 levels were higher in patients with severe acute pancreatitis (13.33±2.11 ng/ml) than matched healthy controls (0.161±0.03 ng/ml) or patients with mild pancreatitis (2.64±0.185 ng/ml) [29]. Soluble RAGE (sRAGE), which lacks the transmembrane and the signaling domain, has the ability to compete with HMGB1 to bind membrane-bound RAGE. In patients with acute pancreatitis, the serum levels of sRAGE inversely correlate with serum levels of HMGB1 [29]. In contrast, serum DNA level may positively correlate with HMGB1 level in patients with acute pancreatitis [29]. Additionally, a significant linear correlation exists between circulating HMGB1 levels and disease score, as well as other serum acute-phase proteins [28]. Interestingly, one study reports that sRAGE, but not HMGB1, is increased in patients with acute pancreatitis [30]. Thus, these findings suggest a complex relationship between HMGB1 and sRAGE in acute pancreatitis.
In several experimental animal models of acute pancreatitis, the level of HMGB1 in the serum and injured organs is significantly increased [31-33]. Inhibition of HMGB1 activity with specific neutralizing antibody from Shino-Test Corporation (Sagamihara, Japan) protects against severe acute pancreatitis and limits lung, liver, and renal dysfunction as well as bacterial translocation to the pancreas [34]. Ethyl pyruvate, a key intermediate of glucose metabolism, has been shown to protect against lethal sepsis partly by inhibition of HMGB1 release [35]. Moreover, early blockade or delayed therapeutic delivery with ethyl pyruvate reduces serum HMGB1 level, ameliorates extra pancreatic organ (e.g., lung, liver, and intestine) injury, and protects rats against established severe acute pancreatitis [36-40]. Danaparoid sodium is a low molecular weight heparinoid with anticoagulant and anti-inflammatory effects. The anti-inflammatory activity of danaparoid sodium against cerulein injection caused acute pancreatitis in rats, which is partly mediated through inhibition of HMGB1 release [41]. In addition, HMGB1-A box [42], pyrrolidine dithiocarbamate [43], antithrombin III [44], honokiol [45], cisplatin [46], and scolopendra sub spinipes mutilans [47] inhibit HMGB1 translocation, release, or activity, which therefore reduces pancreatic injury in severe acute pancreatitis. Thus, extracellular HMGB1 mediates the inflammatory response and organ dysfunction and is likely an effective therapeutic target of acute pancreatitis.
To investigate the role of intracellular HMGB1 in the response to local tissue injury and subsequent systemic inflammatory responses, we recently created mice with pancreas-specific disruption in Hmbg1 (Pdx1-Cre; HMGB1flox/flox, termed CH mice) through using a Cre/ LoxP system [48]. As a control, HMGB1flox/flox mice were termed F/F mice. Both CH and F/F mice are born alive without developmental deficiencies. However, in contrast to F/F mice, the CH mice had more severe experimental acute pancreatitis and very high mortality rates in L-arginine- and cerulein-induced acute pancreatitis [48]. Hematoxylin and eosin stains showed exaggerated death of acinar cells, infiltration of leukocytes, and edema of interstitial issue in the CHmice compared to F/F mice during acute pancreatitis [48]. Serum amylase, the most widely-used marker in the assessment of acute pancreatitis severity, was elevated in CH mice during acute pancreatitis [48]. Moreover, the levels of pancreatic myeloperoxidase (a marker for pancreatic neutrophil recruitment) and serum lactate dehydrogenase (a marker for pancreatic necrosis) were significantly increased in the CH mice compared to F/F mice during acute pancreatitis [48]. The most important step in initiating acute pancreatitis is the activation of intrapancreatic trypsinogen to trypsin [49]. As expected, experimental acute pancreatitis-induced intrapancreatic trypsin activity was also increased in CH mice compared to F/F mice [48]. Binding of HMGB1 to histone and DNA sustains chromosome structure and function. We subsequently observed that loss of HMGB1 in pancreatic acinar cells increased the levels of γ-H2AX (a marker for DNA damage), cleaved-poly ADP ribose polymerase, and cleaved-caspase3 (markers for apoptosis); release of histones/DNA; and activation of inflammatory signaling pathways (e.g., NF-κB and mitogen-activated protein kinases) [48]. Besides HMGB1, histones and DNA are proinflammatory nuclear DAMPs and their release is regulated by oxidative stress. Blocking histone/DNA release has been demonstrated to protect against several inflammation-associated diseases [50]. These findings therefore indicate that intracellular HMGB1 protects against acute pancreatitis through limiting histone and DNA release.
Surprisingly, we observed higher serum HMGB1 in CH mice following experimental acute pancreatitis, suggesting a link between HMGB1 deficiency in local tissue and circulating HMGB1 accumulation [48]. Next, we demonstrated that the source of circulating HMGB1 in CH mice was from histone- and DNA-mediated recruitment and activation of macrophages [48]. In vitro, DNA inhibitor (Dnase I) and histone 3 (H3)-neutralizing antibodies significantly inhibited macrophage migration and HMGB1 release [48]. In vivo, anti-H3 neutralizing antibodies effectively reduced serum HMGB1 levels and reversed the phenotype of CH mice in acute pancreatitis. In addition to blocking histone activity by neutralizing antibodies, inhibition of histone release by antioxidant (e.g., N-acetyl- L-cysteine) also protected CH mice from acute pancreatitis. Finally, we determined whether blocking HMGB1 activity would change the phenotype of CH mice in response to suffering from pancreatitis. Indeed, we demonstrated that anti-HMGB1 neutralizing antibodies prolonged animal survival and decreased serum levels of tissue enzymes (e.g., amylase, lactate dehydrogenase, and myeloperoxidase) and pro-inflammatory cytokines (e.g., tumor necrosis factor-α and interleukin-6) in CH mice following pancreatitis [48]. These findings suggest that the crosstalk between intracellular and extracellular nuclear DAMPs (e.g., HMGB1 and histones) leads to the development of pancreatitis (Figure 1).
HMGB1 is both an architectural nuclear protein and a DAMP. Despite the recent significant advances in understanding the opposing effects of intracellular and extracellular HMGB1 in acute pancreatitis, many questions remain. For example, what regulates the balance between levels of intracellular and extracellular HMGB1? One suggestion is the metabolism production from mitochondrial stress-driven HMGB1 translocation and release [51,52]. Another issue concerns the activity of HMGB1 from different types of death, as HMGB1 redox status may distinguish immunogenic and tolerogenic cell death [53]. Reagents that block HMGB1’srelease and activity ameliorate the symptoms in experimental pancreatitis; therefore, they will probably be tested in clinical trials. Additional large randomized studies are necessary to determine the utility of HMGB1 inhibitor in patients.
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by the National Institutes of Health (NIH) (R01 CA160417 to D.T. and R01CA181450 to H.J.Z/M.T.L) and a 2013 Pancreatic Cancer Action Network-AACR Career Development Award (Grant Number 13-20-25-TANG). The work supporting the findings reviewed in this manuscript was aided by core support from the University of Pittsburgh Cancer Institute (NIH grant P30CA047904).
HMGB1 plays a dual role in acute pancreatitis. Increased stressors cause translocation of HMGB1 from the nucleus to the cytosol and then its release into the extracellular space in pancreatitis. Loss of intracellular HMGB1 in the pancreas leads to nuclear catastrophe and inflammatory nucleosome release, which can further promote inflammatory cell recruitment/activation and HMGB1 release. Finally, increased serum HMGB1 and nucleosomes cause systemic inflammatory response syndrome and multiple organ failure.
Austin Publishing Group is an emerging open access publisher specialising in Science, Technology and Medicine is dedicated to serve the biomedical community through its initiatives. Austin Publishing Group is an academic publisher with 100+ peer reviewed open access journals in various subjects such as biomedical, Pharma, Life Sciences, Environmental, Engineering and Management. Austin Publishing Group publishes Open Access eBooks providing free access to vast scientific literature.