
Review Article
Austin J Vasc Med. 2015; 2(1): 1011.
Hypercholesterolemia Induces Vascular Cell Dysfunction: Molecular Basis for Atherosclerosis
Kerstin Tjaden, Evangelia Pardali and Johannes Waltenberger*
Department of Cardiovascular Medicine, Division of Cardiology, University Hospital Munster, Germany Cells-in-Motion Cluster of Excellence (EXC 1003 – CiM), University of Munster, Germany
*Corresponding author: Johannes Waltenberger, Department of Cardiovascular Medicine, Division of Cardiology, University Hospital Munster, Albert- Schweitzer- Campus 1 - Building A1, 48149 Munster, Germany
Received: July 02, 2015; Accepted: September 10, 2015; Published: September 25, 2015
Abstract
Hypercholesterolemia (HC) is a cardiovascular risk factor characterized by elevated serum levels of lipoproteins. The most important lipoprotein is Low Density Lipoprotein (LDL)-cholesterol. Increased LDL levels have been linked to atherosclerosis and cardiovascular disease. Various modifications such as oxidation or glycation do increase the pathological effects of LDL. Not only the modified but also the native form of LDL is involved in the development of atherosclerosis by influencing various cell types. LDL induces the activation of a number of intracellular pathways leading to increased inflammation as well as hyper activation and dysfunction of vascular cells. In addition, HC induces oxidative stress, which has also been associated with cell dysfunction and atherosclerosis. In this review we discuss the recent advances on HC-induced cellular dysfunction and cardiovascular diseases. These advances provide opportunities for the development of new therapies for cardiovascular diseases.
Keywords: Hypercholesterolemia; Cellular dysfunction; Low density lipoprotein
Abbreviations
ADMA: Asymmetric Dimethyl Arginine; AP: Activator Protein; Apo: Apolipoprotein; CCR: C-C Chemokine Receptor; CVD: Cardiovascular Disease; DM: Diabetes Mellitus; EC: Endothelial Cell; ENOS: Endothelial Nitric Oxide Synthase; ERK: Extracellular- Signal-Regulated Kinase; GTP: Guanosin TriPhosphat; HC: Hypercholesterolemia; HDL: High Density Lipoprotein; HMG- CoA: Hydroxymethylglutaryl-Coenzyme A; IDL: Intermediate Density Lipoprotein; IL: Interleukin; JNK: c-Jun N-terminal Kinase; KO: Knock Out; LDL: Low Density Lipoprotein; LDLR: LDL Receptor; LOX: Lectin - Like Oxidized Low-Density Lipoprotein Receptor; LP(a): Lipoprotein (a); MCP: Monocyte Chemotactic Protein; MI: Myocardial Infarction; mmLDL: minimally modified LDL; MMP: Matrix Metalloproteinase; MPO: Myeloperoxidase; MTP: Microsomal Transfer Protein; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NF- κB: Nuclear Factor Κ-Light-Chain- Enhancer Of Activated B Cells; nLDL; native LDL; NO: Nitric Oxide; NOX: NADPH Oxidase; oxLDL: Oxidized LDL; PAI: Plasminogen Activator Inhibitor; PCSK: Preprotein Convertase Subtilisin Kexin; PI3K: Phosphor Inositide 3-Kinase; PKB: Protein Kinase B; PKC: Protein Kinase C; PTK: Protein Tyrosine Kinase; ROCK: Rho- Associated Protein Kinase; ROS: Reactive Oxygen Species; SMC: Smooth Muscle Cell; SOD: Superoxide Dismutase; SR: Scavenger Receptor; VEGF: Vascular Endothelial Growth Factor; VEGFR: VEGF Receptor; VLDL: Very Low Density Lipoprotein; WHHL: Watanabe Heritable Hyperlipedemic; WT: Wild Type; ZNF: Zinc Finger Protein
Introduction
Atherosclerosis is a chronic inflammatory disease and represents the underlying cause of most Cardiovascular Diseases (CVD). The process of atherogenesis, which is the formation of atheromatous lesions in the vessel wall, is initially associated with increased adhesion of leukocytes to the endothelium activated by irritating stimuli such as hypertension. Recruitment of leukocytes -including monocytes- into the vessel wall is facilitated by inflammatory stimuli like locally produced inflammatory cytokines/chemokines. Monocyte accumulation and their differentiation into macrophages promote plaque growth. Macrophages can endocytose modified lipids and thus develop to foam cells, which further promote plaque progression. Similarly, Smooth Muscle Cells (SMC) migrate into and proliferate within the cap of a plaque. Disruption of the plaque leads to thrombosis, a cause for Myocardial Infarction (MI) or stroke [1]. There are several factors, which have been shown to enhance the risk of (CVD) including Hypercholesterolemia (HC), Diabetes Mellitus (DM), hypertension and smoking. HC is characterized by elevated serum levels of lipoproteins including low density lipoprotein (LDL)- cholesterol. According to the guidelines of the European Society of Cardiology, LDL-cholesterol serum levels above 100mg/dL are considered to be pathologically increased in healthy individuals. For individuals with low risk for CVD, LDL-cholesterol levels should be lower than 70mg/dL [2]. Various studies have linked elevated LDLcholesterol levels to an increased risk of cardiovascular events such as MI [3-6]. For this reason the assessment of LDL-cholesterol levels is part of the standard lipid blood profile in humans [7]. Furthermore, it has been shown, that lipid lowering agents such as statins reduce the risk of vascular events by 20% for approximately 40mg/dL reduction in LDL-cholesterol concentration [8]. Administration of the LDL lowering drug ezetimibe can further enhance the lipid lowering effect with a proven impact on reduced mortality as shown in the IMPROVE-IT trial recently [9]. Ezetimibe inhibits the absorption from cholesterol into the intestine and therefore the delivery to the liver which lowers the intracellular LDL-cholesterol levels [10]. It is well known that HC leads to cellular dysfunction and thereby contributes to the progression of atherosclerosis. Elevated lipids levels lead to enhanced adhesion of leukocytes to endothelial cells (EC) and facilitate infiltration of immune cells into the vessel wall, although the exact mechanisms behind this remains unclear [11]. Furthermore, modified lipids such as oxidized LDL (oxLDL) stimulate and maintain inflammation during atherosclerosis [12]. Due to the important role of HC on CVD development several studies have focused on the clinical effects of HC as a cardiovascular risk in patients and the possibilities to lower lipid levels. This review will focus on effects of HC on cellular function and the consequences for atherosclerosis and CVD.
Lipid Profile
Lipid components and their role in CVD
Total cholesterol consists of different components [13]: High Density Lipoprotein (HDL), Intermediate Density Lipoprotein (IDL), LDL, Very Low Density Lipoprotein (VLDL), Chylomicrons and Triglycerides. Elevated LDL-cholesterol levels are strongly associated with a poor cardiovascular outcome [2]. Interestingly, not only LDL, but also IDL and VLDL have been shown to contribute to atherosclerosis [14]. In contrast to LDL, it has been suggested, that high HDL serum levels are beneficial and that high HDL can be protective against vascular events [15]. HDL contains Apolipoprotein apoB and transports cholesterol from peripheral tissue to the liver where it is cleared from the circulation, a mechanism called reverse cholesterol transport [16]. Nevertheless, recent findings could demonstrate that the oxidized form of HDL exerts cytotoxic effects on monocytes by inducing both, oxidative stress as well as the expression of Matrix Metalloproteinases (MMP), which in turn play an important role in monocyte recruitment during atherosclerosis [17]. Lipoprotein a Lp(a) is present in human serum and consists of two apolipoproteins: B and A. The biochemical structure is similar to that of LDL. In contrast to LDL, LP(a) contains apoA, which is highly glycosylated [18]. Apolipoproteins bind lipids such as cholesterol and form lipoproteins. In that way lipids can be transported within the circulation [19]. LP(a) contributes to inflammation and atherosclerosis by acting as aproinflammatory mediator. LP(a) not only induces the expression of adhesion molecules on EC but also increases chemotaxis of monocytes by inducing the secretion of cytokines. Like LDL, LP(a) can be modified in vivo. Oxidation and glycation of LP(a) may alter its atherogenic potential. Moreover, elevated LP(a) levels in the serum are associated with an increased cardiovascular risk [18].
Biology and pathology of LDL
Cholesterol transport in the human body is mostly executed by LDL, which carries cholesterols to peripheral tissues, where they are taken up. LDL consists of apoB and -like other lipoproteins- not only of cholesterol, but also of phospholipids, triglycerides and cholesteryl esters. The size of the LDL particle can vary due to different amounts of triglycerides and/or cholesteryl esters. LDL- cholesterol is being internalized via the LDL receptor (LDLR), which is absent or modified in familial hypercholesterolemia [20]. As a consequence, LDL cannot be cleared from the circulation and consequently accumulates [21]. By modifying native LDL (nLDL) in various ways, it’s atherogenic and proinflammatory potential can be enhanced. Modifications of LDL are used for in vitro experiments; nevertheless, these processes are relevant in vivo as well, since modifications are taking place within the vascular wall [22]. The most prominent modification is oxidation. There are different approaches to oxidize LDL. The commonly used in vitro method is via metal ions. Copper (Cu2+) or iron (Fe2+) catalyze oxidation of the polyunsaturated fatty acids of LDL, while oxidation via Cu2+ occurs to a stronger degree, i.e. not only the lipid part but also the protein part undergoes oxidation [23]. Lipid hydroperoxides are necessary to initiate this non enzymatic oxidation [24]. The oxidation process can be achieved by cells in vitro and less frequently in vivo as metal ions are not available in such high amounts. Incubation of vascular cells including EC or SMC with LDL leads to its oxidation. This can be catalyzed by different enzymes. One of them is lipoxygenase, which also modifies the polyunsaturated acids of the LDL particle and is expressed mainly in macrophages [22]. The second enzyme catalyzing the oxidation of LDL is myeloperoxidase (MPO), which is preferentially expressed in monocytes and neutrophils. It is known, that MPO is associated with the progression of atherosclerosis. MPO is a heme enzyme and involved in the generation of Reactive Oxygen Species (ROS) such as hypochlorous acid. In turn, ROS contributes to oxidation of the lipid and the protein part of LDL [22]. Nldl is recognized and internalized by LDLR. Also minimally modified or mildly oxidized LDL (mmLDL) can be taken up by this receptor. LDLR is expressed on monocytes, macrophages, EC and SMC. Low concentrations of LDL in the circulation lead to upregulation of the receptor, which induces the clearance of LDL from the circulation. In contrast, high LDL levels lead to downregulation of LDLR expression in order to keep the intracellular LDL concentration continuously on the same level [25]. MmLDL and oxLDL differ in their grade of oxidation. While oxLDL is completely oxidized, mmLDL consists of native parts as well. Scavenger Receptors (SR) such as SR-A, CD36 or oxLDL receptor (LOX)-1 are expressed on macrophages and other vascular cells and bind both mmLDL and oxLDL. In contrast to LDLR the SR are not down regulated in response to high oxLDL concentrations [26]. Another functionally relevant modification of LDL is glycation. The most abundant amino acid in the LDL particle, lysine, undergoes glycation in a non-enzymatic way [27]. It has been demonstrated that LDL in a diabetic environment contains higher amounts of glycated lysine than in non-diabetic conditions [28]. In addition glucose, which is elevated in diabetic conditions, enhances the levels of LDL oxidation [29]. Due to altered density and size of the LDL particle in diabetic patients LDL oxidation is facilitated [30]. They have been shown to be smaller and therefore more dense [31]. Oxidation under high glucose conditions is catalyzed by the enzymes catalase and Superoxide Dismutase (SOD) [30].The modified LDL forms are more pro-atherogenic than the native form as they lead to increased monocyte accumulation and foam cell formation within the plaque [26].
Effects of HC on the vascular system – evidence from in vitro and in vivo models
HC induces cellular dysfunction and inflammation: Endothelial dysfunction is thought to be an early marker for atherosclerosis with changes in cell morphology and oxidative as well as inflammatory status. Atherosclerosis is a chronic inflammatory condition and HC induces several proinflammatory events, which can potentiate atherosclerosis. As mentioned above, oxLDL is a stronger inflammatory stimulus than nLDL. OxLDL activates various cell types such as EC, SMC and leukocytes, and induces the expression of proinflammatory cytokines, adhesion molecules and other inflammatory molecules [13, 32, 33]. Inflammatory cells recruited by oxLDL induced cytokines lead to further LDL oxidation [22]. OxLDL is associated with endothelial dysfunction since it has been shown to trigger endothelial inflammation [34]. LDL induces cytokine production like monocyte chemotactic protein 1(MCP-1), expression of adhesion molecules such as Vascular Cell Adhesion Protein-1 (VCAM-1) and Intercellular Adhesion Molecule-1 (ICAM- 1) and release of MMP. This leads to aberrant activation of EC and EC dysfunction. These effects of oxLDL are partly mediated by LOX- 1 and activation of various signaling cascades such as p38 Mitogen- Activated Protein Kinases (MAPK), extracellular-signal-regulated kinase 1/2 (ERK1/2) MAPK, protein tyrosine kinase (PTK) or nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) in EC [35]. In addition, oxLDL induces MCP-1 secretion and expression of the adhesion molecules ICAM-1 and VCAM-1 in human EC in a LOX-1-dependent manner [36]. Likewise, glycated LDL induces inflammation in human EC by upregulation of MCP-1 and VCAM- 1 in a NF-κB-dependent manner [37]. A recent study could show that oxLDL induced VCAM-1 expression via the NF-κB pathway depends on the interim α5β1 signaling, which is also upregulated by oxLDL in human EC [38]. It has been shown that oxLDL mediates cytokine release in EC by inducing the ERK1/2 pathway, which then regulates histone modifications of the promotor from the ccl2 gene. This effect can be reverted by statins [39]. In addition, LDL increases the permeability of the endothelium, which contributes to increased leukocyte infiltration into the vessel wall [40]. Treatment of human EC with oxLDL and nLDL in combination with cigarette smoke leads to reduction of Nitric Oxide (NO). NO is an important regulator of the vascular tone and vasodilation. Reduced NO bioavailability is a mark of endothelial dysfunction [41]. Exposure of human EC ton LDL or oxLDL induced the expression of Plasminogen activator inhibitor-1(PAI-1), whose activity has been associated with CVD. It has been shown that oxLDL mediates PAI-1 expression via the LOX- 1/Raf-1/ERK1/2 axis [42]. Apoptosis of EC is induced by oxLDL via the ERK1/2 or NF-κB pathway. Also, oxLDL induced EC apoptosis by up regulation of the Fas ligand [43]. Moreover, oxLDL induces the expression of CD40 and its ligand in EC by activating protein kinase C (PKC), which trigger the inflammatory responses in EC [44] (Figure 1).
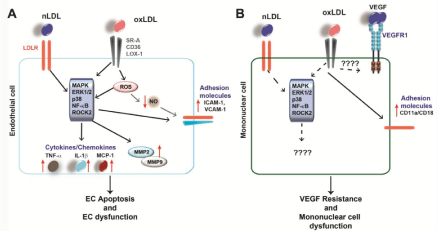
Figure 1: LDL-induced signaling in EC and mononuclear cells
(A) Both nLDL and oxLDL induce intracellular signaling in EC, which leads to enhanced expression of downstream targets such as cytokines, MMP and adhesion
molecules (indicated by red arrows). This leads to EC dysfunction and apoptosis, two mechanisms contributing to atherosclerosis. (B) Both nLDL and oxLDL affect
mononuclear cell function by influencing downstream signaling cascades and target expression, however, the exact molecular mechanisms remain to be fully
elucidated.
Studies with ECs revealed that oxLDL induces Rho-Associated Protein Kinase 2 (ROCK 2) via LOX-1. This activates downstream molecules involved in signaling pathways such as Akt, also known as protein kinase B (PKB), PKC, NF-κB and MAPK [45]. There is further evidence that LDL influences other transcription factors. A study revealed a correlation between the oxLDL/LDL ratio and the expression of the zinc finger protein 580 (ZNF 580) and IL-8 [46]. ZNF 580 has been shown to be a transcription factor involved in MMP2 and Vascular Endothelial Growth Factor (VEGF) regulation [47]. High oxLDL/LDL ratios are found in patients with high numbers of atherosclerotic lesions and are associated with an increased IL-8 expression, which is due to down regulation of ZNF 580 and leads to an increased monocyte adhesion on EC [46]. OxLDL not only induces NF-κB, but also the transcription factor Activator Protein-1 (AP-1), which in turn up regulates the expression of inflammatory genes leading to EC dysfunction [48]. It is known, that ROCK2 influences cell motility, the cytoskeleton, as well as the vascular tone and adhesion [49]. Furthermore, it is also linked to atherosclerosis [50]. Activation of signaling pathways by oxLDL induced ROCK2 further leads to increased chemokine/cytokine expression such as interleukin IL-8 by NF-κB. This in turn recruits monocytes to the inflamed endothelium during atherosclerosis [45]. In addition, EC exposed to LDL show reduced migratory responses towards VEGF probably due to internalization and degradation of VEGF receptor VEGFR2 and reduced VEGF downstream signaling pathways such as phosphatidylinositide 3-kinase (PI3K)/PKB and ERK1/2 [51]. These results suggest, that oxLDL, as well as nLDL have an inhibiting effect on angiogenesis. Similar results have been previously described by us in conjunction with DM. Diabetic monocytes show diminished chemotaxis towards VEGF compared to monocyte migration from healthy individuals, which may explain the reduced angiogenesis (healing) after MI in diabetic patients. This phenomenon is called VEGF resistance [52].Taken together, these finding suggest that oxLDL mediates its pathological effects on EC by activating different signaling cascades.
There are many studies on the role of HC/LDL in EC dysfunction, while the effect of HC on leukocyte/monocyte dysfunction is not so clear yet. Nevertheless, some investigations have been made. While oxLDL induces cytokine secretion in EC to recruit monocytes, it can also act as a chemoattractant for monocytes by itself [53]. oxLDL downregulated the expression of the MCP-1 receptor CCR2 on human monocytic cells THP-1, while it increased the expression of peroxisome proliferator-activated receptor-γ (PPARγ), which is known to regulate the inflammatory response in monocytes/ macrophages [54]. In contrast, chemotaxis of monocytes is increased by up regulation of CCR2 expression by nLDL [55]. It is well known, that macrophages internalize oxLDL via their SR and develop into foam cells, a hallmark of atherosclerosis. Nevertheless, both nLDL and oxLDL exert their pathological effects in different ways on leukocytes [13]. HC affects not only the phenotype and therefore the function, but also proliferation of leukocytes. Studies revealed that HC correlated with increasing numbers of circulating neutrophils and monocytes. It has been shown that HC induces proliferation of both hematopoietic stem and progenitor cells [56]. Moreover, oxLDL increases chemotactic activity of monocytes and their adhesion to endothelial cells by enhancing expression of endothelial adhesion molecules. It has been demonstrated that oxLDL induced the expression of growth factors in EC and macrophages and therefore also the proliferation and migration of SMC [57]. In monocytes/macrophages, oxLDL induces apoptosis by inducing DNA fragmentation [58], whereas another study showed that oxLDL reduced apoptosis by activating the ERK1/2 pathways in the monocytic cell line THP-1 [59]. It is known that the oxLDL receptor CD36 activates MAPK via activation of Src kinases [60]. Also nLDL increases adhesion of leukocytes on the endothelium by inducing the expression of adhesion molecules such as P-selectin in human monocytes. The effects of nLDL are mediated by LDLR [26]. OxLDL induces the toll like receptor (TLR 4/6) hetero dimer assembly via CD36 and therefore the NF-κB pathway andIL-1β secretion [61]. LDL induced the p38 signaling cascade after short term incubation in human platelets, which inhibited the Na+/H+antiport and leads to sensitization [62]. All in all, the findings of the different in vitro studies show that the modified forms of LDL have more severe, pathological effects on vascular cell function compared to the effects of nLDL.
Oxidative stress and cellular dysfunction: Oxidative stress is the imbalance between ROS and mechanisms of a biological system to act against or clear these substances [63]. ROS are not only free radicals (superoxide), but also other oxidants like hydrogen peroxide. Oxidative stress is associated with the progression and presence of CVD [7]. In addition, it has been shown, that oxidative stress leads to cellular dysfunction. ROS could increase the permeability of human EC in vitro [64]. Furthermore, ROS induces cell dysfunction by damaging different cellular components such as proteins, DNA and mitochondria [65]. Three enzymes produce the major amount of ROS, called nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), xanthine oxidase and MPO. HC leads to their up regulation. NOX isoforms catalyze the reaction from superoxide to hydrogen peroxide, whereas NOX2 is the most important isoform in the vascular wall. Xanthine oxidase catalyzes reactions to superoxide and hydrogen peroxide. MPO, which is known to oxidize LDL as described above, catalyzed the production of hypochlorous acid [13]. Enhanced ROS production can lead to oxidative damage of proteins, lipids or DNA. Further oxidative stress induces signaling cascades such as NF-κB or MAPK pathways leading to an up regulation of inflammatory conditions. Also intracellular calcium (Ca2+), which is an important molecule for many signaling processes within the cell, increases due to oxidative stress [7].
Incubation of polymorph nuclear leukocytes with oxLDL but not nLDL increased the intracellular calcium levels [66]. It has also been shown that oxLDL increased NOX activity in both THP-1 and in U937-derived macrophages supporting the fact that HC enhances oxidative stress [67]. Furthermore, it is known that ROS production is induced by oxLDL and LOX-1 interaction leading to upregulation of adhesion molecules in EC and enhanced inflammation [68]. OxLDL induces NOX2 mRNA expression and superoxide production in human EC supporting the role of HC in oxidative stress [69]. Not only oxLDL but also glycated LDL can induce oxidative stress by up regulating the NOX subunits p22phox and NOX4 in EC by activating the NF-κB and the p38 MAPK pathway [70]. Moreover, oxidative stress induced by HC leads to a decrease in NO bioavailability in EC. This is due to increased circulating levels of the endothelial NO synthase (eNOS) inhibitor asymmetric dimethylarginine (ADMA). Since NO as an endothelium-derived relaxing factor is important for maintenance of the vascular tone a reduced NO bioavailability leads to EC dysfunction [71]. Besides its functional impact, ADMA has been shown to be a valid marker for oxidative damage [72]. Taken together, these results suggest that the pathology of HC and LDL respectively is due to a combination of its inflammatory and oxidative properties.
Role of HC in development of atherosclerosis in preclinical models: Animals, such as mice, which have naturally low LDLcholesterol and high HDL levels, do not develop atherosclerosis by themselves [56]. The first successful deletion of a protein in mice resulted in the apoE knockout (KO) mice, which develop spontaneous atherosclerosis, when they are fed with a high fat diet [73]. In addition, this is associated with elevated levels of circulating LDL. It has been shown that dietary plant sterols can prevent HC and atherosclerosis in apoE KO mice [74]. A study with apoE-/- mice fed with Western diet revealed that HC induced monocytosis of monocytes, which were found in lesions at later time points during atherosclerosis and shown to adhere on the endothelium and differentiate to macrophages [75]. In addition, it has been shown that there was increased adhesion of leukocytes on EC in apoE-/- mice after feeding high cholesterol compared to wild type mice. These effects were reversed by simvastatin, a commonly used statin [76]. The effects of HC on the endothelium are due to activation of the NF-κB pathway, since inhibition of this pathway in EC resulted in reduced atherosclerosis in apoE-/- mice fed with a cholesterol rich diet [77]. Another prominent mouse model for studying atherosclerosis is the LDLR KO mouse [78]. LDLR-/- mice develop atherosclerosis as LDL-cholesterol cannot be cleared from the circulation. It has been shown, that overexpression of 15-lipooxigenase in the vascular wall contributes to atherosclerotic lesion formation in these mice [79]. A study with wild-type (WT) and cJun N-terminal kinase (JNK) KO mice fed with high cholesterol diet revealed that JNK deletion protected against endothelial dysfunction. The mechanism is dependent on HC/JNK-induced ROS production [80]. Cholesterol rich diet induced expression of adhesion molecule VCAM-1 in rabbit EC in vivo. This supports the hypothesis that HC induces inflammation [81]. Watanabe Heritable Hyperlipidemic (WHHL) rabbits are a model organism for familial HC since they are characterized by natural LDLR deficiency and high LDL levels in the circulation [82]. It was shown, that hind limb ischemia induced capillary formation was significantly lower in WHHL rabbits compared to control animals [83]. This can be due to defects in VEGF signaling since EC migration and proliferation induced by VEGF is impaired by nLDL in vitro [51]. Studies with hyperlipidemic hamsters demonstrated an increase of nLDL transport in aortic fatty steaks, a preliminary stage of the atherosclerotic plaque, whereas in control animals nLDL was not detectable in these areas [60]. In mice deficient for SR-AI and SR-AII, prominent receptors for oxLDL, a reduced formation of atherosclerotic changes could be seen as well as a reduced number of adherent monocytes, further supporting the role of oxLDL in the progression of atherosclerosis [84].
Role of HC in human CVD: High oxLDL levels are associated with CVD [2]. OxLDL expresses immunogenic epitopes which induce the generation of antibodies against them. These antibodies can be detected in the serum of patients with advanced atherosclerosis and they are used as in vivo markers for LDL oxidation [57]. A study from our group using monocytes from HC patients revealed that the chemotactic response of these cells towards VEGF was attenuated, an observation described as VEGF resistance [85]. Similarly, MCP-1 induced responses were functionally impaired in monocytes from patients with HC [85]. In diabetic patients this is due to increased baseline activation of p38 and PI3K/PKB signaling [86]. This leads to the assumption, that the same mechanism may be active under hypercholesterolemic conditions, which deserves verification. Furthermore, it has been shown, that monocytes from hypercholesterolemic patients treated with simvastatin show reduced release of cytokines such as IL-1β, IL-6 or MCP-1 ex vivo, which leads to reduced activation of these cells [87]. In line with results from preclinical models [56], it has been shown, that there are increased numbers of monocytes and macrophages in patients with familial or non-familial HC [88]. Moreover, monocytes isolated from hypercholesterolemic patients have been shown to produce increasing amounts of ROS, which is due to elevated oxLDL levels [89]. Another study on monocytes from patients with HC revealed that incubation of these cells with nLDL ex vivo led to an increase in expression of the adhesion molecule CD11b compared to monocytes from healthy individuals. The increase was reversed by simvastatin treatment [90]. There are also different studies on the effect of HC on endothelium dependent vasodilation, i.e. means the widening of blood vessels. This is impaired in hypercholesterolemic patients but can be reverted by the use of statins [91-93]. It has also been shown that inhibition of p38 MAPK improves HC-induced impairment of endothelial vasodilation [94]. Platelets from patients with HC are activated and aggregate to a higher degree. NLDL as well as oxLDL have an influence on platelets. Platelets are able to internalize oxLDL via their SR CD36 and thereby induce foam cell formation in macrophages. In that way activated platelets contribute to atherosclerotic plaque formation [95].
Therapeutic Strategies
Lipid lowering strategies: Statins and other lipid lowering agents
The most commonly used medication to treat HC is hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors better known as statins. Their potential to reduce the risk of CVD has been verified in multiple studies [96]. The HMG-CoA reductase catalyzes the last and rate limiting step during cholesterol synthesis. Statins inhibit protein isoprenylation, which plays important roles in signaling processes in the cardiovascular system by interfering with Guanosine Triphosphate (GTP) binding proteins such as Ras, Rho, and Rac. Statins are thought to act beneficial against CVD not only because of their lipid lowering function, but also because of their antioxidant and anti-inflammatory properties, previously referred to as “pleiotropic effects” by reducing oxidative stress [97]. It remains to be demonstrated whether these pleiotropic effects are indeed independent of cholesterol modification.
Statins reduce LDL-cholesterol levels in familial hypercholesterolemic patients up to 50%. Nevertheless, statins have only limited effects on LDL levels in some patient groups [98]. For example, in patients with extremely high baseline LDLcholesterol the treatment with statins could not show a sufficient reduction of these levels [99]. Another problem in daily practice is statin intolerance [100]. Therefore, other lipid lowering strategies are a current topic of research and development. Most of them are used in addition to statins since this is the accepted and established standard therapy for reducing LDL-cholesterol [98]. Among them are cholesterol absorption inhibitors like ezetimibe (IMPROVE-IT) [9]. In combination with statins they have been shown to reduce cardiovascular events by 27% [101]. Another approach exploits microsomal transfer protein (MTP) inhibitors, which inhibit the transport of cholesterol and triglycerides, which in turn stabilizes nascent VLDL particles. They show an efficiency of more than 50% reduction of LDL-cholesterol levels by using high concentrations [102]. Nevertheless, since these inhibitors also increase cholesterol pools in the liver, which in turn leads to fatty liver development only the use of one MTP inhibitor (lomitapide) with less side effects is approved and allowed in humans [102]. Other current developments refer to the possibility to reduce LDL via antisense oligonuclotides. MipomersenTM is used as an antisense therapy and reduces apoB und therefore LDL in human plasma from 20% to 65% [103]. This drug is currently marketed [104].
A novel and very promising LDL lowering therapy is via Preprotein convertase subtilisin kexin-9 (PCSK-9) inhibitors. PCSK-9 is involved in intracellular and extracellular expression of LDLR. Binding of PCSK-9 to LDLR leads to its internalization. It has been proven in different studies and clinical trials that statins lead to upregulation of PCSK [105]. Antibodies against PCSK-9 have been shown to increase LDLR expression and therefore reduce LDL-cholesterol level in human plasma from about 30% to 50% [95]. Although the use of monoclonal antibodies is the most common treatment against PCSK also antisense oligonucleotides and small interfering RNAs have been shown to reduce PCSK and therefore LDL-cholesterol levels [105]. Important clinical trial data are expected soon, especially on Alirocumab, which is being tested in the Odyssey trial [106].
Other anti-inflammatory agents
Statins are thought to be beneficial against CVD not only because of their lipid lowering but also because of their anti-inflammatory potential. Phytosterols are plant derived sterols or stanols [107]. Their structure is similar to the one of cholesterol. They have been shown to reduce not only LDL-cholesterol but also total cholesterol in hypercholesterolemic subjects which is probably due to interaction in micelle formation. Nevertheless, phytosterols also exert antiinflammatory properties [108]. Studies with apoE-KO mice could show a reduced expression of proinflammatory cytokines in atherosclerotic lesions after phytosterol treatment [109]. Clinical data supports these findings [108]. Furthermore, immunization plays a role in therapeutic strategies against CVD. It is known that circulating auto antibodies against LDL exist in human [110]. A study by Nielson and colleagues found a modest inverse correlation between autoantibody titers and the intima-media thickness in healthy people [111]. Monoclonal IgG antibodies against LDL have been shown to reduce atherosclerotic lesion in LDLR deficient mice. However, this antibody failed to achieve the expected results in phase II of a clinical study [110]. By eating moderately, meaning especially reducing the uptake of animal fats and salts and by increasing the consumption of fruits and vegetables, patients with or without cardiovascular problems were able to reduce their weight and LDL-cholesterol levels while improving their HDL levels. Furthermore, also reduced the risk of a cardiovascular event [112]. This shows that not only medication/ drugs can lower lipid levels but also a healthy lifestyle (low fat diet) can contribute to that.
Conclusion
Atherosclerosis is the underlying process of the most important CVDs. HC is one of the most important and modifiable cardiovascular risk factors for atherosclerosis. High LDL plasma levels result in increased inflammation and enhanced oxidative stress as well as cellcell adhesion thereby facilitating the infiltration of leukocytes into the vessel wall and enhances plaque progression. Moreover, HC induces dysfunction of vascular cells, such as EC, SMC and leukocytes by activation intracellular signaling cascades and expression of inflammatory molecules, which further contribute to atherosclerosis. Statins are established drugs to reduce the risk of MI in patients with CVD, as well as to reducing progression of atherosclerosis. Other more potent drugs to reduce HC are currently under development. While much needs to be investigated on the molecular mechanisms that underlie the role of HC in cellular dysfunction and development of atherosclerosis, the recent advances provide ample opportunities for the development of new therapies for CVD.
Acknowledgement
Our studies on the role of growth factor signaling in CVD are supported by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) of the Medical Faculty of the University of Münster, Germany and by the ‘Innovative Medizinische Forschung’ (IMF PA121004) program of the Medical Faculty of the University of Münster, Germany. Furthermore, our studies are supported by Cells-in- Motion Cluster of Excellence (EXC 1003 – CiM), University of Münster, Germany.
References
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011; 473: 317-325.
- Catapano AL, Reiner Z, De Backer G, Graham I, Taskinen MR, Wiklund O, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis. 2011; 217: 012.
- Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007; 370: 1829-1839.
- Langsted A, Freiberg JJ, Tybjaerg-Hansen A, Schnohr P, Jensen GB, Nordestgaard BG. Nonfasting cholesterol and triglycerides and association with risk of myocardial infarction and total mortality: the Copenhagen City Heart Study with 31 years of follow-up. J Intern Med. 2011; 270: 65-75.
- Menotti A, Lanti M, Kromhout D, Blackburn H, Jacobs D, Nissinen A, et al. Homogeneity in the relationship of serum cholesterol to coronary deaths across different cultures: 40-year follow-up of the Seven Countries Study. Eur J Cardiovasc Prev Rehabil. 2008; 15: 719-725.
- Ylä-Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia-Garcia HM, Herrmann J, Hoefer I. Stabilization of atherosclerotic plaques: an update. Eur Heart J. 2013; 34: 3251-3258.
- Peluso I, Morabito G, Urban L, Ioannone F, Serafini M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocr Metab Immune Disord Drug Targets. 2012; 12: 351-360.
- Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet. 2005; 366: 1267-1278.
- Blazing MA, Giugliano RP, Cannon CP, Musliner TA, Tershakovec AM, White JA, et al. Evaluating cardiovascular event reduction with ezetimibe as an adjunct to simvastatin in 18,144 patients after acute coronary syndromes: final baseline characteristics of the IMPROVE-IT study population. Am Heart J. 2014; 168: 205-212.
- Bruckert E, Giral P, Tellier P. Perspectives in cholesterol-lowering therapy: the role of ezetimibe, a new selective inhibitor of intestinal cholesterol absorption. Circulation. 2003; 107: 3124-3128.
- Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991; 88: 1785-1792.
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999; 340: 115-126.
- Stapleton PA, Goodwill AG, James ME, Brock RW, Frisbee JC. Hypercholesterolemia and microvascular dysfunction: interventional strategies. J Inflamm (Lond). 2010; 7: 54.
- Carmena R, Duriez P, Fruchart JC. Atherogenic lipoprotein particles in atherosclerosis. Circulation. 2004; 109: III2-7.
- Barter P: The role of HDL-cholesterol in preventing atherosclerotic disease, Eur Heart J Suppl. 2005; 7.
- Levinson SS, Wagner SG: Implications of reverse cholesterol transport: Recent studies: Clin Chim Acta. 2015; 439C: 154-161.
- Soumyarani VS, Jayakumari N. Oxidized HDL induces cytotoxic effects: implications for atherogenic mechanism. J Biochem Mol Toxicol. 2014; 28: 481-489.
- Malaguarnera M, Vacante M, Russo C, Malaguarnera G, Antic T, Malaguarnera L, et al. Lipoprotein (a) in cardiovascular diseases. Biomed Res Int. 2013; 2013: 650989.
- Mahley RW, Innerarity TL, Rall SC Jr, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. 1984; 25: 1277-1294.
- Varghese MJ. Familial hypercholesterolemia: A review. Ann Pediatr Cardiol. 2014; 7: 107-117.
- Goldstein JL, Brown M. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009; 29: 431-438.
- Yoshida H, Kisugi R. Mechanisms of LDL oxidation. Clin Chim Acta. 2010; 411: 1875-1882.
- Tsimikas S, Miller Y. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr Pharm Des. 2011; 17: 27-37.
- Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992; 13: 341-390.
- Rhainds D, Brissette L. Low density lipoprotein uptake: holoparticle and cholesteryl ester selective uptake. Int J Biochem Cell Biol. 1999; 31: 915-931.
- Gleissner CA, Leitinger N, Ley K. Effects of native and modified low-density lipoproteins on monocyte recruitment in atherosclerosis. Hypertension. 2007; 50: 276-283.
- Steinbrecher UP, Witztum JL. Glucosylation of low-density lipoproteins to an extent comparable to that seen in diabetes slows their catabolism. Diabetes. 1984; 33: 130-134.
- Younis N, Sharma R, Soran H, Charlton-Menys V, Elseweidy M, Durrington PN. Glycation as an atherogenic modification of LDL. Curr Opin Lipidol. 2008; 19: 378-384.
- Kawamura M, Heinecke JW, Chait A. Pathophysiological concentrations of glucose promote oxidative modification of low density lipoprotein by a superoxide-dependent pathway. J Clin Invest. 1994; 94: 771-778.
- Yoshida H, Ishikawa T, Nakamura H. Vitamin E/lipid peroxide ratio and susceptibility of LDL to oxidative modification in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1997; 17: 1438-1446.
- Chait A, Brazg RL, Tribble DL, Krauss RM. Susceptibility of small, dense, low-density lipoproteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern B. Am J Med. 1993; 94: 350-356.
- Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. 2014; 5: 927-946.
- Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013; 2013: 152786.
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109: III27-32.
- Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006; 69: 36-45.
- Hashimoto K, Kataoka N, Nakamura E, Tsujioka K, Kajiya F. Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis. 2007; 194: e9-17.
- Toma L, Stancu CS, Sanda GM, Sima AV. Anti-oxidant and anti-inflammatory mechanisms of amlodipine action to improve endothelial cell dysfunction induced by irreversibly glycated LDL. Biochem Biophys Res Commun. 2011; 411: 202-207.
- Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW: alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014; 34: 1362-1373.
- Dje N'Guessan P, Riediger F, Vardarova K, Scharf S, Eitel J, Opitz B,et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler Thromb Vasc Biol. 2009; 29: 380-386.
- Pirro M, Bagaglia F, Paoletti L, Razzi R, Mannarino MR. Hypercholesterolemia-associated endothelial progenitor cell dysfunction. Ther Adv Cardiovasc Dis. 2008; 2: 329-339.
- Steffen Y, Vuillaume G, Stolle K, Roewer K, Lietz M, Schueller J, et al. Cigarette smoke and LDL cooperate in reducing nitric oxide bioavailability in endothelial cells via effects on both eNOS and NADPH oxidase. Nitric Oxide. 2012; 27: 176-184.
- Sangle GV, Zhao R, Shen GX. Transmembrane signaling pathway mediates oxidized low-density lipoprotein-induced expression of plasminogen activator inhibitor-1 in vascular endothelial cells. Am J Physiol Endocrinol Metab. 2008; 295: 16.
- Sata M, Walsh K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J Clin Invest. 1998; 102: 1682-1689.
- Li D, Mehta JL. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circ Res. 2009; 104: 566-568.
- Mattaliano MD, Wooters J, Shih HH, Paulsen JE. ROCK2 associates with lectin-like oxidized LDL receptor-1 and mediates oxidized LDL-induced IL-8 production. Am J Physiol Cell Physiol. 2010; 298: C1180-1187.
- Hoffmann CJ, Hohberg M, Chlench S, Maroski J, Drab M, Siegel G, et al. Suppression of zinc finger protein 580 by high oxLDL/LDL-ratios is followed by enhanced expression of endothelial IL-8. Atherosclerosis. 2011; 216: 103-108.
- Sun HY, Wei SP, Xu RC, Xu PX, Zhang WC. Sphingosine-1-phosphate induces human endothelial VEGF and MMP-2 production via transcription factor ZNF580: novel insights into angiogenesis. Biochem Biophys Res Commun. 2010; 395: 361-366.
- Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med. 2014; 70: 117-128.
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003; 4: 446-456.
- Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci. 2006; 27: 97-104.
- Jin F, Hagemann N, Brockmeier U, Schäfer ST, Zechariah A, Hermann DM. LDL attenuates VEGF-induced angiogenesis via mechanisms involving VEGFR2 internalization and degradation following endosome-trans-Golgi network trafficking. Angiogenesis. 2013; 16: 625-637.
- Waltenberger J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem Soc Trans. 2009; 37: 1167-1170.
- McMurray HF, Parthasarathy S, Steinberg D. Oxidatively modified low density lipoprotein is a chemoattractant for human T lymphocytes. J Clin Invest. 1993; 92: 1004-1008.
- Han KH, Chang MK, Boullier A, Green SR, Li A, Glass CK, et al. Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor gamma. J Clin Invest. 2000; 106: 793-802.
- Han KH, Chen Y, Chang MK, Han YC, Park JH, Green SR, et al. LDL activates signaling pathways leading to an increase in cytosolic free calcium and stimulation of CD11b expression in monocytes. J Lipid Res. 2003; 44: 1332-1340.
- Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013; 24: 129-136.
- Maiolino G, Rossitto G, Caielli P, Bisogni V, Rossi GP, Calò LA. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm. 2013; 2013: 714653.
- Hardwick SJ1, Hegyi L, Clare K, Law NS, Carpenter KL, Mitchinson MJ, et al. Apoptosis in human monocyte-macrophages exposed to oxidized low density lipoprotein. J Pathol. 1996; 179: 294-302.
- Namgaladze D, Kollas A, Brüne B. Oxidized LDL attenuates apoptosis in monocytic cells by activating ERK signaling. J Lipid Res. 2008; 49: 58-65.
- Stancu CS, Toma L, Sima AV. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012; 349: 433-446.
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010; 11: 155-161.
- Nofer JR, Noll C, Feuerborn R, Assmann G, Tepel M. Low density lipoproteins inhibit the Na+/H+ antiport in human platelets via activation of p38MAP kinase. Biochem Biophys Res Commun. 2006; 340: 751-757.
- Halliwell B, Cross CE. Oxygen-derived species: their relation to human disease and environmental stress. Environ Health Perspect. 1994; 102 Suppl 10: 5-12.
- Shasby DM, Lind SE, Shasby SS, Goldsmith JC, Hunninghake GW. Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood. 1985; 65: 605-614.
- Williams RS. Canaries in the coal mine: mitochondrial DNA and vascular injury from reactive oxygen species. Circ Res. 2000; 86: 915-916.
- van Tits LJ, Hak-Lemmers HL, Demacker PN, Stalenhoef AF, Willems PH. Oxidized low-density lipoprotein induces calcium influx in polymorphonuclear leukocytes. Free Radic Biol Med. 2000; 29: 747-755.
- Nguyen-Khoa T, Massy ZA, Witko-Sarsat V, Canteloup S, Kebede M, Lacour B, et al. Oxidized low-density lipoprotein induces macrophage respiratory burst via its protein moiety: A novel pathway in atherogenesis? Biochem Biophys Res Commun. 1999; 263: 804-809.
- Dunn S, Vohra RS, Murphy JE, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. The lectin-like oxidized low-density-lipoprotein receptor: a pro-inflammatory factor in vascular disease. Biochem J. 2008; 409: 349-355.
- Rueckschloss U, Galle J, Holtz J, Zerkowski HR, Morawietz H. Induction of NAD(P)H oxidase by oxidized low-density lipoprotein in human endothelial cells: antioxidative potential of hydroxymethylglutaryl coenzyme A reductase inhibitor therapy. Circulation. 2001; 104: 1767-1772.
- Sangle GV, Zhao R, Mizuno TM, Shen GX. Involvement of RAGE, NADPH oxidase, and Ras/Raf-1 pathway in glycated LDL-induced expression of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Endocrinology. 2010; 151: 4455-4466.
- Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflugers Arch. 2010; 459: 807-816.
- Böger RH, Bode-Böger SM, Tsao PS, Lin PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J Am Coll Cardiol. 2000; 36: 2287-2295.
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992; 258: 468-471.
- Kapourchali FR, Surendiran G, Chen L, Uitz E, Bahadori B, Moghadasian MH. Animal models of atherosclerosis. World J Clin Cases. 2014; 2: 126-132.
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007; 117: 195-205.
- Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, Trocha SD, et al. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001; 103: 2598-2603.
- Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008; 8: 372-383.
- Sanan DA, Newland DL, Tao R, Marcovina S, Wang J, Mooser V, et al. Low density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein(a). Proc Natl Acad Sci USA. 1998; 95: 4544-4549.
- Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, et al. Over expression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2000; 20: 2100-2105.
- Osto E, Matter CM, Kouroedov A, Malinski T, Bachschmid M, Camici GG, et al. c-Jun N-terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation. 2008; 118: 2073-2080.
- Li H, Cybulsky M, Gimbrone MA, Libby P. Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol. 1993; 143: 1551-1559.
- Kondo T, Watanabe Y. A heritable hyperlipemic rabbit. Jikken Dobutsu. 1975; 24: 89-94.
- Van Belle E, Rivard A, Chen D, Silver M, Bunting S, Ferrara N, et al. Hypercholesterolemia attenuates angiogenesis but does not preclude augmentation by angiogenic cytokines. Circulation. 1997; 96: 2667-2674.
- Stephen SL, Freestone K, Dunn S, Twigg MW, Homer-Vanniasinkam S, Walker JH, et al. Scavenger receptors and their potential as therapeutic targets in the treatment of cardiovascular disease. Int J Hypertens. 2010; 2010: 646-929.
- Czepluch FS, Bergler A, Waltenberger J. Hypercholesterolaemia impairs monocyte function in CAD patients. J Intern Med. 2007; 261: 201-204.
- Tchaikovski V, Olieslagers S, Böhmer FD, Waltenberger J. Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation. 2009; 120: 150-159.
- Krysiak R, Okopien B. Different effects of simvastatin on ex vivo monocyte cytokine release in patients with hypercholesterolemia and impaired glucose tolerance. J Physiol Pharmacol. 2010; 61: 725-732.
- Fadini GP, Simoni F, Cappellari R, Vitturi N, Galasso S, Vigili de Kreutzenberg S2, Previato L2. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis. 2014; 237: 805-808.
- Vasconcelos EM, Degasperi GR, de Oliveira HC, Vercesi AE, de Faria EC, Castilho LN. Reactive oxygen species generation in peripheral blood monocytes and oxidized LDL are increased in hyperlipidemic patients. Clin Biochem. 2009; 42: 1222-1227.
- Serrano CV Jr, Pesaro AE, de Lemos JA, Rached F, Segre CA, Gomes F et al. Native LDL-cholesterol mediated monocyte adhesion molecule over expression is blocked by simvastatin. Cardiovasc Drugs Ther. 2009; 23: 215-220.
- Chowienczyk PJ, Watts GF, Cockcroft JR, Ritter JM. Impaired endothelium-dependent vasodilation of forearm resistance vessels in hypercholesterolaemia. Lancet. 1992; 340: 1430-1432.
- O'Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997; 95: 1126-1131.
- Heitzer T, Ylä-Herttuala S, Luoma J, Kurz S, Münzel T, Just H, et al. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation. 1996; 93: 1346-1353.
- Cheriyan J, Webb AJ, Sarov-Blat L, Elkhawad M, Wallace SM, Mäki-Petäjä KM, et al. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011; 123: 515-523.
- Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014; 46: e99.
- Illingworth DR1, Tobert JA. A review of clinical trials comparing HMG-CoA reductase inhibitors. Clin Ther. 1994; 16: 366-385.
- Tousoulis D, Psarros C, Demosthenous M, Patel R, Antoniades C, Stefanadis C. Innate and adaptive inflammation as a therapeutic target in vascular disease: the emerging role of statins. J Am Coll Cardiol. 2014; 63: 2491-2502.
- Wierzbicki AS, Viljoen A, Hardman TC, Mikhailidis DP. New therapies to reduce low-density lipoprotein cholesterol. Curr Opin Cardiol. 2013; 28: 452-457.
- Wierzbicki AS, Perera D, Ewang-Emukowhate M. Dyslipidaemia: what's around the corner? Clin Med. 2014; 14: s41-44.
- Yan AT, Yan RT, Tan M, Hackam DG, Leblanc KL, Kertland H, Tsang JL. Contemporary management of dyslipidemia in high-risk patients: targets still not met. Am J Med. 2006; 119: 676-683.
- Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011; 377: 2181-2192.
- Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007; 356: 148-156.
- Kastelein JJ, Wedel MK, Baker BF, Su J, Bradley JD, Yu RZ, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006; 114: 1729-1735.
- Khoo B: Genetic therapies to lower cholesterol. Vascul Pharmacol. 2014; 24: 00208-00200.
- Sahebkar A, Watts GF. New LDL-cholesterol lowering therapies: pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin Ther. 2013; 35: 1082-1098.
- Moriarty PM, Jacobson TA, Bruckert E, Thompson PD, Guyton JR, Baccara-Dinet MT, et al. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK9, in statin-intolerant patients: design and rationale of Odyssey Alternative, a randomized phase 3 trial. J Clin Lipidol. 2014; 8: 554-561.
- Ostlund RE Jr. Phytosterols in human nutrition. Annu Rev Nutr. 2002; 22: 533-549.
- Othman RA, Moghadasian MH. Beyond cholesterol-lowering effects of plant sterols: clinical and experimental evidence of anti-inflammatory properties. Nutr Rev. 2011; 69: 371-382.
- Nashed B, Yeganeh B, HayGlass KT, Moghadasian MH: Antiatherogenic effects of dietary plant sterols are associated with inhibition of proinflammatory cytokine production in Apo E-KO mice. J Nutr. 2005; 135: 2438-2444.
- Chyu KY, Shah PK. Advances in immune-modulating therapies to treat atherosclerotic cardiovascular diseases. Ther Adv Vaccines. 2014; 2: 56-66.
- McLeod O, Silveira A, Fredrikson GN, Gertow K, Baldassarre D, Veglia F, et al. Plasma autoantibodies against apolipoprotein B-100 peptide 210 in subclinical atherosclerosis. Atherosclerosis. 2014; 232: 242-248.
- Chaves G, Britez N, Munzinger J, Uhlmann L, Gonzalez G, Oviedo G, et al. Education to a Healthy Lifestyle Improves Symptoms and Cardiovascular Risk Factors - AsuRiesgo Study. Arq Bras Cardiol. 2015; 104: 347-355.