
Review Article
Austin Alzheimers J Parkinsons Dis. 2014;1(2): 8.
Late Phase Cell-Cycle Proteins in Postmitotic Neurons: Relation to Alzheimer’s Disease?
Bajic VP1*, Stanimirovic J1, Obradovic M1,Zivkovic L2,Milicevic Z1 and Spremo-Potparevic B2
1Vinca Institute of Nuclear Sciences, University of Belgrade, Serbia
2Department of Pharmacy, University of Belgrade, Serbia
*Corresponding author: Bajic VP, Laboratory of Radiobiology and Molecular Genetics, Vinca Institute for Nuclear Sciences, University of Belgrade, P.O Box 522, 11001 Belgrade.
Received: August 27, 2014; Accepted: September 20, 2014; Published: September 25, 2014
Abstract
Cell cycle re-entry has become a well established model of neuropathogenesis in Alzheimer’s disease (AD). We and others have demonstrated expression of early phase cell cycle-related proteins in the vulnerable neurons in AD. Evidence that this represents a bona fide mitotic event is verified by the observation that DNA replication does in fact occur in these cells. Notably, the relatively early occurrence of cell cycle events in AD suggests that a mitotic cell cycle related mechanism may play a pivotal role in the disease. Still, a number of features of the cell cycle re-entry phenotype have remained elusive to the role of ectopic protein expression in the process of neuronal cell death. Late phase cell cycle proteins regulate separation and segregation of chromosomes. The centromere region is crucial to this process. Until recently there has been no data on the role of proteins controlling the centromere region in postmitotic neurons. This new data suggests that cohesin complexes that mediate sister–chromatid cohesion in dividing cells may also contribute to gene regulation in postmitotic cells. Therefore, centromere-cohesin proteins may play a secondary role in the cell, i.e. one that is independent of their role in cohesion and chromosome segregation. Evidence demonstrating that instability of centromere-cohesion dynamics in the early phases of the cell cycle which coincide with re-entry alteration of cortical neurons enables the possibility to further elucidate initial processes leading to AD.
Keywords: Alzheimer’s Disease; Cell-cycle reentry; Centromere; Cohesin
Abbreviations
AD: Alzheimer’s disease; APC: Anaphase Promoting Complex; APLP2: Amyloid Precursor-Like Protein 2; APP: Amyloid Precursor Protein; Aβ: Amyloid-β; Cdc20: Cell-Division Cycle Protein 20; Cdh1: Cdc20-Homologue 1; CDK: Cyclin Dependent Kinase; GSK3β: Glycogen Synthase Kinase 3 β; NFT: Neuro Fibrillary Tangle; NR: N-methyl-D-aspartate Receptor; PCD: Premature Centromere Division; PSD-95: Postsynaptic Density-95; PTTG: Pituitary Tumor Transforming Gene; SAC: Spindle Assembly Checkpoint; Scc: Sister Chromatide Cohesion Protein; SMC: Structural Maintenance of Chromosome; TIM-1: Timeless 1
Introduction
The canonical markers of AD are extracellular senile plaque composed primarily of fibrilar amyloid-β (Aβ) peptides, and the intracellular neurofibrillary tangle (NFT), a mass of irregularly folded proteins composed mainly of hyperphosphorylated tau protein. The causes and consequences of both Aβ and tau accumulation are a primary focus in the field. In particular, the relationship between tau and Aβ on other mechanisms known to be involved in disease pathogenesis has garnered considerable attention. In this regard changes involving cell cycle dynamics appear to be centrally involved. Indeed, the intracelullar accumulation of highly phosphorylated tau is linked to the cell cycle and cell cycle dependent kinases [1]. Cell senescence, oxidative stress, misregulated apoptosis are important factors in the pathogenesis of AD [1,2] and are influenced by aberrations in the cell cycle dynamics and, in particular, telomere length. Notably, the link between cell cycle related events and apoptosis is becoming increasingly recognized in neurodegeneration [3] and it is apparent from studies of neuronal cultures that Aβ mediated cell death [4], only occurs if cells re-entry into a mitotic state [5]. The post-mitotic, quiescent state of adult neurons is long standing dogma in neuroscience. However, there is accumulating evidence that neurons re-enter the cell division cycle in AD [4, 6-18]. In mammalian cells, this mitotic re-entry depends on extracellular signals, namely on the balance between mitogenic stimuli and differentiating factors [4,19,20]. Sequential expression, activation and degradation of cyclin/Cyclin Dependent Kinase (CDK) complexes drive the cell cycle, and their regulation is achieved via mechanisms of transcription, phosphorylation, proteolysis, and association with CDK Inhibitors (CDKI) [21]. G0/G1 phase transfer in the cell cycle is triggered by the presence of cyclin D/Cdk4 and Cdk6 complex [22]. When DNA replication is completed, the cyclin A/Cdk2 complex enables transition from the S to the G2 phase (S/G2) of the cell cycle. For the cell to enter the G2 phase of mitosis (G2/M), degradation of cyclin A/Cdk 2 complex and expression of cyclin B which activates Cdk2 must take place [22]. Two major observations have helped establish the cell cycle hypothesis for AD neurodegeneration. First, activation of cell cycle regulators generally precedes formation of degenerative lesions such as NFTs, and the regulators and downstream effectors eventually become incorporated into NFTs [23]. Therefore, it has been postulated that cell-cycle regulators initiate and mediate the neurodegenerative process . Second, with the exception of karyokinesis and cytokinesis, markers from every phase of the cell cycle, early and late phase (metaphase-anaphase transition) which are consequently presented by duplicated chromosomes (tetraploidy) [24-27], aneuploidy [27-35], bi-nuclear cells [36] and Premature Centromere Division (PCD) [37] have been found in degenerating neurons (Table 1). The present phenotypes represent a cell ahead of the S phase. Still, the AD cell goes into a G2/M block and eventually dies through the process of apoptosis. Except for chromosome 21 that has been found to be aneuplodogenic and in a tetraploidy state in neurons affected by AD [24-35], chromosome X has been recently reported to show instability reported as aneuploidy [38] and skewing [39]. Both, aneuploidy and skewing are related to the centromere instability phenotype, or PCD [37] (Table 1).
![]()
Cell cycle phase
Cell cycle markers
Accompanied Phenotype
G0-G1
Mitogenic/trophic factors and receptors, downstream signaling, increased pRb and E2F1
None
G1-S
Cdk4; cyclins E and A, PCNA, p105, Cdc25A,CDK 5
None
G2-M
Cdc2; cyclin B1; Cdc25B; Wee1, Cdk7,PLK1, increased phosphorylation of nucleolin, RNA pol II; Wee1; Cdc25 A and B, Cdk 5, Cohesin, APC, Cdk11
tetraploidy;
aneuploidy;
bi-nucleation,
premature centromere division
Abbreviations: CDK: Cyclin Dependent Kinase; Cdc: Cell-division Cycle Protein; PCNA: Proliferating Cell Nuclear Antigen; pRb: Retinoblastoma Protein; PLK 1: Polo Kinase 1; RNA pol II: RNA Polymerase
* The phenotypes represent only that the AD cell has passed the G1-S phase of the cell cycle but went into a G2/M block as no AD cell has ever completed a full cell cycle. The proteins reported in the G2/M phase of Table 1, are ectopically expressed in AD and have been extensively been reviewed elsewhere.
Table 1: Expression of cell cycle markers with accompanied phenotype in the AD brain.
This has led to the hypothesis that vulnerable neurons re-enter the cell cycle and proceed through the S phase, but then abort somatic division and eventually degenerate. These observations have raised the following questions: What triggers this final cycle? When does this occur? Are neurons truly postmitotic, or they do cycle very, very slowly? To answer these questions and to fully understand the AD cell, we have postulated a new hypothesis that says “A new shift toward understanding the regulation of centromere-cohesion associated proteins in the processes of cell cycle re-entry in the AD brain is needed”. This means that in a postmitotic cell the function of proteins have changed, once they functioned for the cell to dived and now they function in a postmitotic cell in order to actively retain the interphase state, quiescent state. Cell cycle regulators have diversified roles in postmitotic neurons, i.e. Cdc25 A and B, Wee 1 are constitutively active in normal postmitotic neurons of the brain [40-43]. Cyclins of the S phase and B1 are commonly expressed in postmitoic neurons of middle aged and elderly humans [1,9,20,25,28,43-45] as are cyclin H [46], and anaphase promoting complex (APC) [47-54]. Wendt et al [55] showed that cohesin has a transcriptional role during the G1 phase and in postmitotic neuronal cells. This shows that chromosome segregation, transcriptional regulation and repair of DNA double strand breaks require the cohesin protein complex [56,57] (Figure 1).
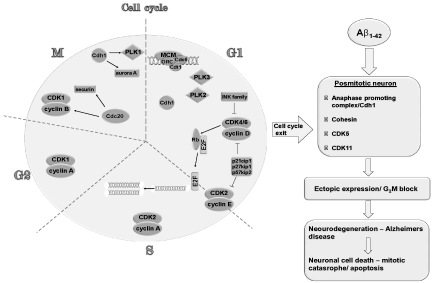
Figure 1: A schematic representation showing a normal neuronal cell cycle and excocytotxic action of Aß on the postmitotic neuron altering crucial late phase cell cycle proteins. This figure represents a generalized interaction between late phase cell cycle proteins *(Cohesin, APC/Cdh 1, CDK 5, CDK 11) that have an active role in the maintenance of a postmitotic state of a neuron and early phase cell cycle proteins that are represented on the left. Extensive research have been done on early phase proteins and are not the subject of this present paper. Mitotic signals from Aß lead to our view to an abortive cell cycle in which ectopic expression of the late phase cell cycle proteins in coordination with early phase proteins induce a G2/M block leading cell death.
Alzheimer Disease-Associated Proteins and the Cell Cycle are intimately Linked
Tau phosphorylation
The major protein component of the NFT, the major intracellular pathology of AD, is a highly phosphorylated form of the microtubule associated protein tau [58]. The increased phosphorylation of tau destabilizes microtubule dynamics and, consequently, results in neuronal dysfunction [58-60]. However, a near identical phosphorylation of tau, driven by CDKs, also occurs when cells are mitotically active [1,25,44]. Interestingly, in AD, cell cycle protein expression precedes the appearance of phosphorylated tau [61] indicating a possible cause-effect relationship. In this regard, CDKs localized in neurons in AD are known to phosphorylate tau in in vitro assays in a manner similar to that found in AD in vivo [1,25,44]. Ittner et al [62] showed that tau as an axonal protein has a dendritic function in postsynaptic targeting of Fyn, i.e. reduced levels of postsynaptic Fyn and instability of N-methyl-D-aspartate receptor/postsynaptic density-95 complexes (NR/PSD-95) in tau -/- mice prevent Aβ toxicity. Recently we found that astrin a substrate of glycogen synthase kinase 3 β (GSK3β) is increased in AD brains (results not shown). Astrin has a role in cohesion [63] and it is a nonmotor associated protein essential for the cell cycle progression, sister chromatid cohesion and for the completion of mitosis [63]. GSK3 β interacts with and phosphorylates astrin resulting in targeting astrin to the microtubules and kinetochores. GSK3β is one of the most implicated tau kinases and its hyperphosphorylation in AD [64].
Amyloid-β
The major protein component of senile plaques is a 4.2 kDa polypeptide Aβ, which is derived from a larger precursor - Amyloid Precursor Protein (APP) encoded on chromosome 21. Attesting to the importance of this protein, mutations in the APP gene are linked to the inevitable onset of familial AD. Given the role of mitotic re-entry in AD, it is notable that APP is upregulated secondary to mitogenic stimulation [65] and that APP metabolism is regulated by cell cycle-dependent changes [66]. Interestingly, Aβ itself is mitogenic in vitro [23,67] and therefore may play a direct role in the induction and/or propogation of cell cycle-mediated events in AD. In this regard, recent seminal work by Karl Herrup’s group found that in APP transgenic mouse models, including Tg2576, which are Aβ rich [68], the cell cycle is also induced and leads to DNA duplication (i.e. neurons enter S phase). How centromere-cohesion associated proteins are related to APP and tau is not known. One view is that proteins that have a role in cohesion and when altered produce a phenotype that is commonly found in AD (cohesion imbalance or PCD), and may have a secondary role in the re-entry neuronal processes in AD [69]. Presenilin (PS) proteins were recently suggested to play a role in chromosome segregation [29,31,70]. These proteins are functionally related to APP, with a possible topological interaction in the cell membrane [70]. One might therefore speculate that they are also functionally connected with amyloid precursor-like protein 2 (APLP2) [71,72]. Rassoulzadegan et al [73] postulated a different function for an APP family protein. The same gene which was designated Aplp2 (due to sequence similarities between APP and the encoded protein) had been originally described under the name Cdebp. It is a DNA-binding protein that recognizes the sequence [A/G]TCAC[G/A]TG, which is identical to the CDEI element of the yeast centromere. Immunocytochemical analysis localized the protein to discrete spots in the interphase nucleus, and a series of convergent observations then suggested a role in the replication and/or segregation of genomic DNA. Protein binding to a CDEI motif in the genome of bovine papillomavirus type 1 was found to be required for maximal efficiency of replication of the viral DNA in transfected cells and for its subsequent episomal maintenance in stable transformants. A strong inhibitory effect on early development had been observed upon microinjection into fertilized mouse eggs of double-stranded oligonucleotides containing the CDEI sequence, and after treatment of one-cell embryos with antisense oligonucleotides. In both instances, the development was arrested before the blastocyst stage, with the characteristic accumulation of abnormal nuclear structures and DNA contents [73], suggestive of a possible role for the protein in DNA replication and/or chromosome segregation. Many sporadic and familial AD patients, including those carrying PS mutations, exhibit a defect in chromosome segregation that leads to aneuploidy, including trisomy 21, in many cells throughout the body [24,25,29,74,75]. This finding is intriguing because individuals with full trisomy of 21 (Down syndrome) all develop AD pathology at a very early age [29,31,76-78]. Boeras et al [31] found that PS-1 transfected cells indicate that PS cause abnormal chromosome missegregation by altering the structure/function of the mitotic cells. Also, others showed that a number of APP over expressing lines, including the Tg2576 line proposed herein, show increases in cell cycle protein by immunocytochemistry and chromosome duplication by fluorescence in situ hybridization [25]. Therefore, established models in vivo (Tg2576) and in vitro models that display alterations in cell cycle, tau phosphorylation, Aβ, neurodegeneration, and cognitive parameters, (i.e. cardinal features of AD), show that chromosome-centromere instability is not merely a correlative manifestation of the disease process but is possibly a contributing factor to AD.
Secondary Role of Cohesion and Related Proteins in AD: a hypothesis
Temporal instability seen as PCD of vulnerable chromosomes in neuronal and peripheral blood cells in AD show that cohesion/ cohesin is altered resulting in the impairment of the secondary roles of cohesin in the neuronal cell which may lead to more instability and consequently cell death [67]. The centromere and chromatid arms are held by a protein called cohesin [79,80]. Cohesin contains two subunits from the Structural Maintenance of Chromosome (SMC) family, Smc1 and Smc3, and two non-SMC subunits, sister chromatide cohesion protein 1 (Scc1) also called Rad21, and Scc3. Cohesin is loaded onto chromosomes before S phase and establishes cohesion between the duplicated chromosomes (sister chromatids) during DNA replication. This regulated linkage is released in preparation for chromosome segregation through a well-defined mechanism that involves the phosphorylation and proteolytic cleavage of the non-SMC cohesin subunit Scc1. In contrast, the mechanisms that underlie the loading and assembly of cohesin onto chromosomes are poorly understood [79]. The cohesion complex can influence gene expression [81], regulation of time directly through the clock gene paralogue Timeless 1 (TIM-1) [82], homologue repair [83] and check point control [84]. AD patients show centrally and peripherally circardian dysfunction, leading to increased aneuploidy [29,32,33,35] and to neuronal cell death - the basis on which symptoms of dementia in AD reside. Cohesion is maintained during interphase and the cell cycle by a number of proteins [80] and the most abundant proteins in sister chromatid separation are securins, separins and cohesins. Securin accumulation during interphase and their binding to separin prevents premature separin activation. During the normal cell cycle, APC eventually degrades securin, thus activating separin to facilitate chromosome segregation. Accordingly, loss of securin, the inhibitor of separin function, would lead to constitutive separin activation, enabling cohesion removal from heterochromatin regions resulting in Premature Centromere Separation (PCD) [85]. Mice lacking securin show aberrant cell cycle progression and PCD [86]. The Pituitary Tumor Transforming Gene (PTTG), a securin, was cloned initially from a rat pituitary tumor. Overexpression of PTTG activates the expression of p53 and modulates its function, with this action of PTTG being mediated through the regulation of c-myc expression. PTTG also up-regulates the activity of the bax promoter and increases the expression of bax through modulation of p53 expression. PTTG can regulate apoptosis in both a p53-dependent and a p53-independent manner. Structural homology suggested that PTTG may be a mammalian securin and this has been confirmed by its involvement in regulating sister chromatid separation during mitosis [86,87]. Also, Jallepali et al [87] show that PTTG in mice and securin appears to be critically for the maintenance of chromosome stability and cell cycle progression. By using a hybrid model for exploring chromosome 21 instability and RNAi processing, Fukagawa et al [84] showed that dicer-deficient cells express mitotic defects and that many cells died in interphase by apoptosis. The data showed that cells died of aberrant localization of cohesion protein Rad21 with chromosomes expressing PCD and Bub1 inactivation [84]. Inactivation of Bub1 shows that the check point pathway has a defect. If the mitotic checkpoint is defective, the cells should progress through anaphase. However, cells cannot enter anaphase because the sister chromatids and centromeres have separated prematurely and the chromosomes are not aligned at the metaphase plate leaving the cells with multiple spindles. Leland et al [88] showed that abnormal Bub1 function results in a premature separation of chromosomes into separated sister chromatides, a mechanism that has been proposed to be involved in the genesis of the majority of human germ cell aneuploidy. We conclude that a number of Spindle Assembly Checkpoint (SAC) proteins [89,90] cohesion proteins [91,92] and a specific cyclin, Cdk11 [93] when inhibited can express centromere instability phenotype in various cells. The findings that cohesion instability leads to late phase phenotype in affected neurons or PCD [35] and tetraploidy [25] are bound to an alteration of the SAC control. This is strengthen by the notion that mitotic catastrophe can occur after failed mitosis, during the activation of the polyploidy checkpoint, in a partially p53-dependent fashion. DNA damage repair that is p53 dependent is known to be altered in AD [94]. The spindleassembly checkpoint and the postmitotic checkpoint (DNA rereplication in these cells is prevented by a p53-dependent mechanism that arrests cells in postmitotic G1 phase, often referred to as the postmitotic checkpoint) have a fundamental role in safeguarding chromosomal stability [95]. Based on our current knowledge of PCD we postulate that centromere instability of neuronal cells increase over time leading to mitotic catastrophe and premature cell death. Mitotic catastrophe which has been recognized as one of the earliest events in neuronal degeneration, may in fact be sufficient to initiate the neurodegenerative cascade [96].
Anaphase promoting complex in postmitotic neurons
Anaphase-promoting complex or cyclosome is an E3 ubiquitin protein ligase that together with cell-division cycle protein 20(Cdc20) and Cdc20-homologue 1 (Cdh1) targets mitotic proteins for degradation by the proteosome and functions in regulating cell cycle transitions in proliferating cells and has, as revealed recently novel roles in postmitotic neurons [48-54]. APC/C triggers the onset of anaphase and inducing the destruction of sister chromatid cohesion by ubiquinating a protein called securin. Securin is an inhibitory chaperone of a thiol protease called separase. Activated separase cleaves Scc1 that holds sister chromatides together (see Figure 2).
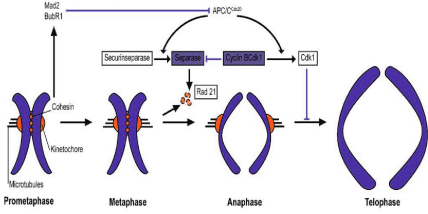
Figure 2: A schematic presentation on the role of APC in the separation and segregation of a chromosome (published in [107]).
APC is regulated by its activator Cdh-1, which is abundantly found in postmitotic neurons. Cdh 1-APC and Cdc20-APC have emerged as key regulators of diverse aspects of neuronal connectivity, from axon to dendrite morphogenesis to synapse remolding and development [47-54]. This shows that APC has multiple roles at different cellular locations and appear not to be connected to cell cycle regulation. Auila et al [51] show that APC may have a role in suppression of cyclin B in postmitotic neurons, thus preventing terminally differentiated neurons to re-enter the cell cycle. This role may be impaired in AD, i.e. it provides an explanation for the mechanism of cyclin B1 reactivation in AD [25,48].
Wirth et al [97] showed that inactivation of APC subunit Apc2 in quiescent hepathocytes may stimulate re-entry into the cell cycle, without any mitogenic stimulus, leading to hepatic failure. They concluded: “This finding implies that the APC has a crucial role in quiescent hepatocytes, namely, to restrain their re-entry into the cell cycle. The APC could perform this function either by suppressing the production of extracellular mitogens or more directly by suppressing accumulation of intracellular proteins capable of promoting proliferative growth. The authors conclude that abolition of the APC causes a major change in the state of quiescent hepatocytes such that they are more readily stimulated to embark on cell proliferation. Crucial to post mitotic maintenance of neurons except APC is CDK5. CDK 5 is an atypical cyclin that inhibits the cell cycle in order to keep neurons in their postmitotic state. Transfer of CDK 5 out of the nucleus to the cytoplasm induces neurodegeneration. APP alfa , a neuroprotective APP cleavage product is found to regulate expression and activity of CDK 5 in primary cortical neurons [98]. CDK 5 dysregulation has been associated with amyloid-beta. This CDK 5/Abeta association triggers the neuronal abortive cell cycle [99]. CDK 5 is associated to APC through phosphorylation and thus inactivation of Cdh1, thus regulating the post mitotic state [100]. These observations possibly link APC ectopic expression through a CDK 5 dependent mechanism and to the beginning of neuronal cell cycle re-entry and to associated phenotypes (Table 1).
Cdk11 and cohesion
Cyclin-dependent kinase 11 (CDK11) mRNA produces a 110- kDa protein (CDK11p110) throughout the cell cycle, a 58-kDa protein (CDK11p58) that is specifically translated from an internal ribosome entry site and expressed only in the G2/M phase of the cell cycle, and a 46-kDa protein (CDK11p46) that is considered to be apoptosis specific. CDK11 is required for sister chromatid cohesion and the completion of mitosis. Cdk11 which is activated only in the G2/M phase of the cell cycle has been found to be adversely affected by AD [70]. A number of researchers have found that, when inhibited, Cdk11 can have a devastating effect on the animal or cell culture phenotype. Corresponding knockout mice do not survive and cells depleted of Cdk11 show cytogenetic abnormality, such as PCD. Dongli et al [93] showed that depletion of Cdk11 delocalizes the cohesion protein Rad21. A number of authors have reported that cells with depleted Rad21 show centromere instability (PCD phenotype). Rad 21 has been demonstrated to cleavage by caspase proteins to trigger sister chromatid separation during apoptotic signaling [101]. Rad21 may show dependence on the presence of the swedish mutation in M17 cells and the TG 2576 mouse (data not shown). Using immunoflourescence Rad21 and Cdk11 are found to localize in the nuclei of M17 cells and M17 APP and M17 swedish mutant type. Based on western blot and densitometry, Rad21 expression is decreased in M17 APPswed mutant. The decrease is also found in Tg+ mice. This correspondence indicates that cohesion in postmitotic neurons is linked to expression of APP and that decreased levels of Rad21 may also show impaired DNA repair in the AD brain. In respect to these results, increased expression of Cdk11 has been found in cellular processes [70] may indicate that Cdk11 is linked to APP signal transduction processing in neuronal synapses [70]. The PCD phenotype has lead us to the conclusion that the AD cell has impaired regulation of cohesion and may have activated the cell cycle checkpoint showing that the AD cell goes ahead of its time. Still, mechanisms that regulated the AD cell does not favor abrogation of the G2/M phase, i.e. not one AD cell has successfully completed its cycle.
Cell Cycle Alterations in Post-Mitotic Neurons May Lead to Cell Death in Alzheimer Disease
Although various mitotic markers are upregulated in vulnerable neurons in AD and these neurons successfully replicate DNA (i.e. proceeds to S phase), no evidence of actual mitosis has ever been found. This suggests that they are arrested at a point (s) prior to the actual event of cellular division. However, it is well known that once cyclin A is expressed, the arrested cells lack the ability to return to G0 and therefore must either complete the cycle or die. Therefore, given the lack of evidence for successful completion of the cell cycle, it is likely that the re-activation of cell cycle machinery in post-mitotic neurons leads to their death. How this is achieved is not currently known. A new hypothesis [69] has been postulated in which alteration of centromere dynamics may play an intrinsic role in cell death by mitotic catastrophe. Ogawa et al [96] state that mitotic catastrophe, recognized as one of the earliest events in neuronal degeneration, may, in fact, be sufficient to initiate the neurodegenerative cascade. Mitotic catastrophe is a poorly defined type of cell death linked to the abnormal activation of cyclin B/Cdk1. Here we propose that a conflict in cell cycle progression or DNA damage can lead to mitotic catastrophe, provided that cell cycle checkpoints are inhibited (in particular the DNA structure checkpoints and the SAC. SAC is required for mitotic catastrophe induced by DNA-damaging agents [102]. Thus, checkpoint proteins which have a role in maintaining the postmitotic state in neurons also influence intrinsic resistance to stress [103]. Oxidative stress and DNA damage in neurons and astrocytes have been found to be elevated in AD [104-106]. Chromosome stability is compromised in vulnerable neurons of the AD brain. To elucidate expression patterns of centromere-cohesion regulated proteins which include the SAC could reveal an underlying mechanism of instability which expresses itself not only in neuronal cells of the frontal cerebral cortex but also in the peripheral blood lymphocytes of AD patients.
Conclusion
Aside from its two characteristic pathologies, Aβ and NFTs, AD demonstrates a mitotically active phenotype. These affected cells show features of activation of the beginning of a full cell cycle, from activation of cyclins and CDKs to activation of cell cycle checkpoint control proteins. Replication of DNA in AD neurons (cyclin B and CDK 1) has been successfully correlated with various cytogenetic alterations of neuronal chromosomes, and one corresponds to the occurrence of tetraploidy in AD neurons [25,36]. Other groups have found chromosome instability, PCD of chromosome X in cortical neurons [37], bi-nuclear neuronal cells in AD brains. Regardless, these cytogenetic alterations not only show that replication has occurred in these cells, but that a number of proteins which constitute the centromere complex have been misregulated meaning that the post mitotic state has been compromised. By elucidating expression patterns of centromere regulated proteins including those of the SAC, we may be able to reveal an underlying mechanism of instability. This review in which we elaborate a role of late phase cell cycle proteins in maintaining a stable post mitotic state of neurons may enable to find new biomarkers that could lead us to better prevention strategies and possible new pathways for pharmacological intervention. Ultimately, the curious mitotic alterations in neurons and peripheral blood lymphocytes of AD, believed to be a result of aberrant cohesion processing, is a characteristic that warrants further investigation.
References
- McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, et al. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta. 2007; 1772: 467-472.
- Franco S, Blasco MA, Siedlak SL, Harris PL, Moreira PI, Perry G, et al. Telomeres and telomerase in Alzheimer's disease: epiphenomena or a new focus for therapeutic strategy? Alzheimers Dement. 2006; 2: 164-168.
- Nagy Z. Cell cycle regulatory failure in neurones: causes and consequences. Neurobiol Aging. 2000; 21: 761-769.
- Raina AK, Zhu X, Smith MA. Alzheimer's disease and the cell cycle. Acta Neurobiol Exp (Wars). 2004; 64: 107-112.
- Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, et al. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000; 275: 11553-11560.
- Arendt T, Holzer M, Gärtner U, Brückner MK. Aberrancies in signal transduction and cell cycle related events in Alzheimer's disease. J Neural Transm Suppl. 1998; 54: 147-158.
- Arendt T, Rödel L, Gärtner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport. 1996; 7: 3047-3049.
- Smith TW, Lippa CF. Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995; 54: 297-303.
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997; 17: 3588-3598.
- Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996; 132: 413-425.
- Nagy ZS, Esiri MM. Apoptosis-related protein expression in the hippocampus in Alzheimer's disease. Neurobiol Aging. 1997; 18: 565-571.
- Nagy ZS, Smith MZ, Esiri MM, Barnetson L, Smith AD. Hyperhomocysteinaemia in Alzheimer's disease and expression of cell cycle markers in the brain. J Neurol Neurosurg Psychiatry. 2000; 69: 565-566.
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997; 150: 1933-1939.
- McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999; 52: 525-527.
- Gerst JL, Raina AK, Pirim I, McShea A, Harris PL, Siedlak SL, et al. Altered cell-matrix associated ADAM proteins in Alzheimer disease. J Neurosci Res. 2000; 59: 680-684.
- Harris PL, Zhu X, Pamies C, Rottkamp CA, Ghanbari HA, McShea A, et al. Neuronal polo-like kinase in Alzheimer disease indicates cell cycle changes. Neurobiol Aging. 2000; 21: 837-841.
- Zhu X, Raina AK, Boux H, Simmons ZL, Takeda A, Smith MA. Activation of oncogenic pathways in degenerating neurons in Alzheimer disease. Int J Dev Neurosci. 2000; 18: 433-437.
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000; 59: 880-888.
- Lavoie JN, Rivard N, L'Allemain G, Pouysségur J. A temporal and biochemical link between growth factor-activated MAP kinases, cyclin D1 induction and cell cycle entry. Prog Cell Cycle Res. 1996; 2: 49-58.
- Vincent I, Pae CI, Hallows JL. The cell cycle and human neurodegenerative disease. Progress in cell cycle research. 2003; 5: 31-41.
- Reed SI. G1/S regulatory mechanisms from yeast to man. Prog Cell Cycle Res. 1996; 2: 15-27.
- Graña X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995; 11: 211-219.
- cDonald DR, Bamberger ME, Combs CK, Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998; 18: 4451-4460.
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001; 21: 2661-2668.
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003; 23: 2557-2563.
- Frade JM, López-Sánchez N. A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75 (NTR). Cell Cycle. 2010; 9: 1934-1941.
- Arendt T, Brückner MK, Mosch B, Lösche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010; 177: 15-20.
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci. 2007; 27: 6859-6867.
- Granic A, Padmanabhan J, Norden M, Potter H. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell. 2010; 21: 511-520.
- Zivkovic L1, Spremo-Potparevic B, Djelic N, Bajic V. Analysis of premature centromere division (PCD) of the chromosome 18 in peripheral blood lymphocytes in Alzheimer disease patients. Mech Ageing Dev. 2006; 127: 892-896.
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer's presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging. 2008; 29: 319-328.
- Thomas P, Fenech M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer's disease. Mutagenesis. 2008; 23: 57-65.
- Iourov IY, Vorsanova SG, Liehr T, Yurov YB. Aneuploidy in the normal, Alzheimer's disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis. 2009; 34: 212-220.
- Zekanowski C, Wojda U. Aneuploidy, chromosomal missegregation, and cell cycle reentry in Alzheimer's disease. Acta Neurobiol Exp (Wars). 2009; 69: 232-253.
- Arendt T, Mosch B, Morawski M. Neuronal aneuploidy in health and disease: a cytomic approach to understand the molecular individuality of neurons. Int J Mol Sci. 2009; 10: 1609-1627.
- Zhu X, Siedlak SL, Wang Y, Perry G, Castellani RJ, Cohen ML, et al. Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol. 2008; 34: 457-465.
- Spremo-Potparevic B, Zivkovic L, Djelic N, Plecas-Solarovic B, Smith MA, Bajic V. Premature centromere division of the X chromosome in neurons in Alzheimer's disease. J Neurochem. 2008; 106: 2218-2223.
- Yurov YB, Vorsanova SG, Liehr T, Kolotii AD, Iourov IY. X chromosome aneuploidy in the Alzheimer's disease brain. Molecular cytogenetics. 2014; 7: 20.
- Bajic V, Mandusic V, Stefanova E, Bozovic A, Davidovic R, Zivkovic L, et al. Skewed X-Chromosome Inactivation in Women Affected by Alzheimer's Disease. J Alzheimers Dis. 2014.
- Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer's disease. Am J Pathol. 2000; 157: 1983-1990.
- Tomashevski A, Husseman J, Jin LW, Nochlin D, Vincent I. Constitutive Wee1 activity in adult brain neurons with M phase-type alterations in Alzheimer neurodegeneration. J Alzheimers Dis. 2001; 3: 195-207.
- Vincent I, Bu B, Hudson K, Husseman J, Nochlin D, Jin L. Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer's disease. Neuroscience. 2001; 105: 639-650.
- Hernandez-Ortega K, Ferrera P, Arias C. Sequential expression of cell-cycle regulators and Alzheimer's disease-related proteins in entorhinal cortex after hippocampal excitotoxic damage. J Neurosci Res. 2007; 85: 1744-1751.
- Bonda DJ, Evans TA, Santocanale C, Llosá JC, Viña J, Bajic V, et al. Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging (Albany NY). 2009; 1: 382-388.
- Ye W, Blain SW. S phase entry causes homocysteine-induced death while ataxia telangiectasia and Rad3 related protein functions anti-apoptotically to protect neurons. Brain. 2010; 133: 2295-2312.
- Jin K, Nagayama T, Chen J, Stetler AR, Kawaguchi K, Simon RP, et al. Molecular cloning of a cell cycle regulation gene cyclin H from ischemic rat brain: expression in neurons after global cerebral ischemia. J Neurochem. 1999; 73: 1598-1608.
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci USA. 1999; 96: 11317-11322.
- Almeida A, Bolaños JP, Moreno S. Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J Neurosci. 2005; 25: 8115-8121.
- Teng FY, Tang BL. APC/C regulation of axonal growth and synaptic functions in postmitotic neurons: the Liprin-alpha connection. Cell Mol Life Sci. 2005; 62: 1571-1578.
- Stegmüller J, Bonni A. Moving past proliferation: new roles for Cdh1-APC in postmitotic neurons. Trends Neurosci. 2005; 28: 596-601.
- Aulia S, Tang BL. Cdh1-APC/C, cyclin B-Cdc2, and Alzheimer's disease pathology. Biochem Biophys Res Commun. 2006; 339: 1-6.
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, et al. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell. 2009; 136: 322-336.
- Puram SV, Kim AH, Bonni A. An old dog learns new tricks: a novel function for Cdc20-APC in dendrite morphogenesis in neurons. Cell Cycle. 2010; 9: 482-485.
- Yang Y, Kim AH, Bonni A. The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Curr Opin Neurobiol. 2010; 20: 92-99.
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008; 451: 796-801.
- Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, et al. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008; 321: 566-569.
- Heidinger-Pauli JM, Unal E, Koshland D. Distinct targets of the Eco1 acetyltransferase modulate cohesion in S phase and in response to DNA damage. Mol Cell. 2009; 34: 311-321.
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010; 7: 656-664.
- Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007; 113: 501-511.
- Buxton GA, Siedlak SL, Perry G, Smith MA. Mathematical modeling of microtubule dynamics: insights into physiology and disease. Prog Neurobiol. 2010; 92: 478-483.
- Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, et al. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem Int. 2009; 54: 84-88.
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010; 142: 387-397.
- Thein KH, Kleylein-Sohn J, Nigg EA, Gruneberg U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J Cell Biol. 2007; 178: 345-354.
- Chen KL, Yuan RY, Hu CJ, Hsu CY. Amyloid-ß peptide alteration of tau exon-10 splicing via the GSK3ß- SC35 pathway. Neurobiol Dis. 2010; 40: 378-385.
- Ledoux S, Rebai N, Dagenais A, Shaw IT, Nalbantoglu J, Sekaly RP, et al. Amyloid precursor protein in peripheral mononuclear cells is up-regulated with cell activation. J Immunol. 1993; 150: 5566-5575.
- Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 1994; 13: 1114-1122.
- Pyo H, Jou I, Jung S, Hong S, Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998; 9: 871-874.
- Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007; 8: 368-378.
- Bajic VP, Spremo-Potparevic B, Zivkovic L, Djelic N, Smith MA. Is the time dimension of the cell cycle re-entry in AD regulated by centromere cohesion dynamics? Biosci Hypotheses. 2008; 1: 156-161.
- Bajic VP, Su B, Lee HG, Kudo W, Siedlak SL, Zivkovic L, et al. Mislocalization of CDK11/PITSLRE, a regulator of the G2/M phase of the cell cycle, in Alzheimer disease. Cell Mol Biol Lett. 2011; 16: 359-372.
- Flood FM, Cowburn RF, Johnston JA. Presenilin-, amyloid precursor protein and amyloid precursor-like protein 2 mRNA levels in human superior frontal cortex during aging. Neuroscience letters. 1997; 235: 17-20.
- Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D'Adamio L. Processing of beta-amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J Biol Chem. 2002; 277: 44195-44201.
- Rassoulzadegan M, Yang Y, Cuzin F. APLP2, a member of the Alzheimer precursor protein family, is required for correct genomic segregation in dividing mouse cells. The EMBO journal. 1998; 17: 4647-4656.
- Migliore L, Testa A, Scarpato R, Pavese N, Petrozzi L, Bonuccelli U. Spontaneous and induced aneuploidy in peripheral blood lymphocytes of patients with Alzheimer's disease. Hum Genet. 1997; 101: 299-305.
- Migliore L, Botto N, Scarpato R, Petrozzi L, Cipriani G, Bonuccelli U. Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer disease patients. Cytogenetics and cell genetics. 1999; 87: 41-46.
- Potter H, Geller LN. Alzheimer's disease, Down's syndrome, and chromosome segregation. Lancet. 1996; 348: 66.
- Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis. 1999; 6: 167-179.
- Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996; 3: 16-32.
- Nasmyth K. How do so few control so many? Cell. 2005; 120: 739-746.
- Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009; 43: 525-558.
- Dorsett D. Roles of the sister chromatid cohesion apparatus in gene expression, development, and human syndromes. Chromosoma. 2007; 116: 1-13.
- Chan RC, Chan A, Jeon M, Wu TF, Pasqualone D, Rougvie AE, et al. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature. 2003; 423: 1002-1009.
- McKay MJ, Troelstra C, van der Spek P, Kanaar R, Smit B, Hagemeijer A, et al. Sequence conservation of the rad21 Schizosaccharomyces pombe DNA double-strand break repair gene in human and mouse. Genomics. 1996; 36: 305-315.
- Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, et al. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol. 2004; 6: 784-791.
- Nasmyth K, Peters JM, Uhlmann F. Splitting the chromosome: cutting the ties that bind sister chromatids. Science. 2000; 288: 1379-1385.
- Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol. 2001; 15: 1870-1879.
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, et al. Securin is required for chromosomal stability in human cells. Cell. 2001; 105: 445-457.
- Leland S, Nagarajan P, Polyzos A, Thomas S, Samaan G, Donnell R, et al. Heterozygosity for a Bub1 mutation causes female-specific germ cell aneuploidy in mice. Proc Natl Acad Sci U S A. 2009; 106: 12776-12781.
- Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, Ito E, et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 2006; 140: 358-367.
- Kienitz A, Vogel C, Morales I, Muller R, Bastians H. Partial downregulation of MAD1 causes spindle checkpoint inactivation and aneuploidy, but does not confer resistance towards taxol. Oncogene. 2005; 24: 4301-4310.
- Hoque MT, Ishikawa F. Cohesin defects lead to premature sister chromatid separation, kinetochore dysfunction, and spindle-assembly checkpoint activation. J Biol Chem. 2002; 277: 42306-42314.
- Díaz-Martínez LA, Giménez-Abién JF, Clarke DJ. Regulation of centromeric cohesion by sororin independently of the APC/C. Cell Cycle. 2007; 6: 714-724.
- Hu D, Valentine M, Kidd VJ, Lahti JM. CDK11(p58) is required for the maintenance of sister chromatid cohesion. J Cell Sci. 2007; 120: 2424-2434.
- Hooper C, Meimaridou E, Tavassoli M, Melino G, Lovestone S, Killick R. p53 is upregulated in Alzheimer's disease and induces tau phosphorylation in HEK293a cells. Neurosci Lett. 2007; 418: 34-37.
- Chan YW, On KF, Chan WM, Wong W, Siu HO, Hau PM, et al. The kinetics of p53 activation versus cyclin E accumulation underlies the relationship between the spindle-assembly checkpoint and the postmitotic checkpoint. J Biol Chem. 2008; 283: 15716-15723.
- Ogawa O, Zhu X, Lee HG, Raina A, Obrenovich ME, Bowser R, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer's disease: a mitotic catastrophe? Acta Neuropathol. 2003; 105: 524-528.
- Wirth KG, Ricci R, Gimenez-Abian JF, Taghybeeglu S, Kudo NR, Jochum W, et al. Loss of the anaphase-promoting complex in quiescent cells causes unscheduled hepatocyte proliferation. Genes & development. 2004; 18: 88-98.
- Hartl D, Klatt S, Roch M, Konthur Z, Klose J, Willnow TE, et al. Soluble alpha-APP (sAPPalpha) regulates CDK5 expression and activity in neurons. PLoS One. 2013; 8: e65920.
- Lopes JP, Oliveira CR, Agostinho P. Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle. 2009; 8: 97-104.
- Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolaños JP, Almeida A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008; 27: 2736-2745.
- Chen F, Kamradt M, Mulcahy M, Byun Y, Xu H, McKay MJ, et al. Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J Biol Chem. 2002; 277: 16775-16781.
- Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 2004; 23: 6548-6558.
- Olsen A, Vantipalli MC, Lithgow GJ. Checkpoint proteins control survival of the postmitotic cells in Caenorhabditis elegans. Science. 2006; 312: 1381-1385.
- Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010; 59: 290-294.
- Gustaw-Rothenberg K, Lerner A, Bonda DJ, Lee HG, Zhu X, Perry G, et al. Biomarkers in Alzheimer's disease: past, present and future. Biomark Med. 2010; 4: 15-26.
- Myung NH, Zhu X, Kruman II, Castellani RJ, Petersen RB, Siedlak SL, et al. Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age (Dordr). 2008; 30: 209-215.
- Bajic VP, Bonda DJ, Zivkovic L, Milicevic Z, Plecas-Solarovic B, Xiongwei Z, et al. The Role of Centromere Cohesion and Associated Proteins in Alzheimer's Disease: A Relation to Aneuploidy? De Rossi S, Bianchi F, editors. In: Aneuploidy: Etiology, Disorders and Risk Factors. 1st edn. Nova Science Publisher, Inc. 2012; 77-91.