
Review Article
Austin Alzheimers J Parkinsons Dis. 2014;1(2): 7.
Cycle on Wheels: Is APP Key to the AppBp1 Pathway?
Chen Y1,2*, Neve RN4, Zheng H3, Griffin WST1,2, Barger SW1,2 and Mrak RE5
1Department of Geriatrics, University of Arkansas for Medical Sciences, USA
2Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, USA
3Huffington Center on Aging, Baylor College of Medicine, USA
44Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, USA
5Department of Pathology, University of Toledo Health Sciences Campus, USA
*Corresponding author: Chen Y, Department of Geriatrics and Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Received: September 02, 2014; Accepted: September 29, 2014; Published: September 30, 2014
Abstract
Alzheimer’s disease (AD) is the gradual loss of the cognitive function due to neuronal death. Currently no therapy is available to slow down, reverse or prevent the disease. Here we analyze the existing data in literature and hypothesize that the physiological function of the Amyloid Precursor Protein (APP) is activating the AppBp1 pathway and this function is gradually lost during the progression of AD pathogenesis. The AppBp1 pathway, also known as the neddylation pathway, activates the small ubiquitin-like protein nedd8, which covalently modifies and switches on Cullin ubiquitin ligases, which are essential in the turnover of cell cycle proteins. Here we discuss how APP may activate the AppBp1 pathway, which downregulates cell cycle markers and protects genome integrity. More investigation of this mechanism-driven hypothesis may provide insights into disease treatment and prevention strategies.
Keywords: APP; Alzheimer’s disease; Ubiquitination; Neddylation; Cell cycle
Abbreviations
APP: Amyloid Precursor Protein; AICD: APP Intracellular Domain; Aβ: β-Amyloid; NAE: Nedd8 Activating Enzyme, i.e. AppBp1 and Uba3 heterodimer; CCM: Cell Cycle Marker; C57: APP’s C-Terminal 57 amino acids; AppBp1: APP-Binding Protein-1, also known as APP-BP1 or NAE1; DDB1: UV-Damaged, DNA-Binding protein 1; DCAF: DDB1-Cul4-associated factor
Introduction
The Amyloid Precursor Protein (APP) is central to understanding Alzheimer’s disease (AD) pathogenesis due to its genetic, biochemical and neuropathological connections with AD. First, APP is the source of β-amyloid (Aβ), a major component of senile plaques in AD brains. Secondly, Genetic mutations of APP cause familial AD [1,2]. Furthermore, an increase in APP gene dosage also causes Aβ deposition and related dementia [3-5]. In the case of trisomy of chromosome 21, AD neuropathology develops universally due to an extra copy of APP [6]. Therefore, elucidating the function of APP may give insights into disease prevention and treatment strategies.
APP is a Type-1 transmembrane receptor [7-10], which is cleaved by multiple proteases. Sequential cleavage by β-secretase BACE1 at the extra-cellular domain [11-13] followed by γ-secretase cleavage inside the membrane [14-16] generates secreted APP N-terminal fragment sAPPβ, Aβ peptides of various length, and APP intracellular domain (AICD) [17-19]. Alternatively, cleavage of APP by the α-secretase within the Aβ domain [20,21] followed by γ-secretase cleavage generates sAPPα, p3, and AICD. Despite intensive studies of APP [22-24] and its cleavage products, the function of APP remains poorly understood.
Molecular basis for APP in cell cycle regulation
In an effort to understand APP as a potential signaling receptor,at least 18 proteins have been identified to bind AICD [25-27]. Among them is APP-binding protein-1 (AppBp1), which binds APP’s C-terminal 57 amino acids (C57) [28]. Co-immunoprecipitation experiments further defined AppBp1’s binding site to two segments of C57: one is adjacent to the membrane including three lysine residues and the other in the C-terminal 31 amino acids [29,30] (Figure 1A). The function of AppBp1 was unknown when it was cloned as an APPbinding protein. The significance of the interaction emerged when AppBp1 was discovered as a cell cycle protein. The first evidence was obtained from hamster ts41 cells, which harbor a temperaturesensitive mutant of AppBp1’s homologue, ts41 [31,32]. At the nonpermissive temperature of 40oC, ts41 cells undergo apoptosis after successive DNA synthesis without cell division [32]. Transfection of the human homologue AppBp1 into ts41 cells restores normal cell cycle at the non-permissive temperature [33]. These data establish AppBp1 as a key player in cell cycle progression across the S-M checkpoint.
The cell cycle is tightly regulated by ubiquitination through an enzymatic cascade that transfers ubiquitin to selected proteins for proteasomal degradation. The first clue to the function of AppBp1 in the cell cycle is that it is highly homologous to the N-terminus of the ubiquitin-activating enzyme Uba1 [28] (Figure 1B). However, AppBp1 lacks the C-terminal conserved cysteine residue necessary for the formation of a thioester bond with ubiquitin [28,34]. AppBp1 was soon shown to bind Uba3, which is highly homologous to the C-terminus of Uba1 and has the corresponding active site cysteine [35,36] (Figure 1B). Together, AppBp1 and Uba3 form a bipartite Nedd8 Activating Enzyme (NAE) for the ubiquitin-like protein nedd8 (see reviews [37,38]). In the enzymatic cascade that activates nedd8, AppBp1 is upstream of Uba3 since Uba3 is not able to restore ts41 cell growth when AppBp1 is inactivated by non-permissive temperature [33]. Mutation of Uba3 in C. elegans also profoundly affects mitosis, presumably by affecting the same nedd8-activation pathway [39].

Figure 1: APP binds to AppBp1 in a region that mediates adenylation and nedd8 binding to NAE. A. C-terminal sequence of APP695 showing the C-terminus of Aβ40 and Aβ42, lysine residues (purple) that can be neddylated and the AppBp1 binding sites in APP AICD region (yellow lines). B. Diagram of conserved domains between NAE and Uba1. The APP-binding site, AppBp1 (145-251) as indicated was identified by Chow et al. [28]. The domains in AppBp1, Uba3, and Uba1 are drawn based on Schulman and Harper’s review [38].
By now it is understood that NAE activates nedd8 by an ATPdependent mechanism analogous to the activation of ubiquitin by Uba1 [38,40,41] (Figure 2). Activation of nedd8 results in the fully loaded NAE complex containing two nedd8 molecules, covalently bound nedd8 thioester and nedd8-AMP that occupies the NAE adenylation domain. This form of NAE activates the transfer of nedd8 to the nedd8-conjugating enzyme by a transthiolation reaction. Eventually, nedd8 is covalently attached to a Cullin in a lysine residue in the conserved Cullin domain. Neddylation of the Cullin induces a conformational change that couples the ubiquitin-charged ubiquitinconjugating enzyme with the substrate for ubiquitination. Many Cullin substrates are involved in cell cycle control and often elevated in cancer cells [42]. Due to its role in activating Cullin ubiquitin ligase, the neddylation pathway is being targeted for cancer growth inhibition [41].
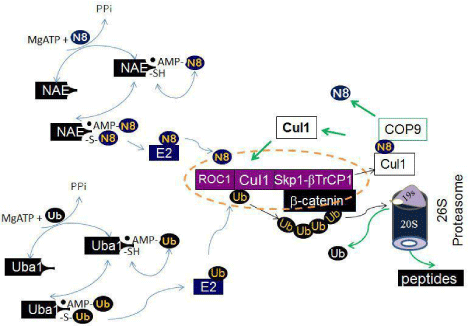
Figure 2: Schematic of neddylation, ubiquitination and proteasomal degradation. In the neddylation pathway, Mg++ATP and nedd8 bind NAE nucleotide binding site, react to yield nedd8-AMP, and release inorganic pyrophosphate. Nedd8-AMP then react with the thiol of the NAE active site cysteine, form the NAE-nedd8 thioester, and release AMP. A second ATP and nedd8 bind AppBp1/Uba3 to form nedd8-AMP, resulting in the fully loaded complex containing two nedd8 molecules, covalently bound nedd8 thioester and nedd8-AMP that occupies the NAE adenylation domain. This form of NAE activates the transfer of nedd8 to the conjugating enzyme by a transthiolation reaction. Subsequently, nedd8 is covalently attached to a Cullin in a lysine residue in the conserved Cullin domain. Neddylation of the Cullin activates the Cullin ubiquitin ligase. In the ubiquitination pathway, Uba1 functions similarly to NAE, but activates ubiquitin, which is ligated to target proteins when Cullin ubiquitin ligase are activated by neddylation. A Cul1 ubiquitin ligase complex (purple) known as SCF complex, is given as an example. The SCF complex consists of the zinc-finger protein, which binds to the C-terminus of Cul1, the adaptor protein Skp1, which binds to the N-terminus of Cul1. The substrate β-catenin is recruited by a specific F-box protein βTrCP1. The ubiquitin chain is recognized by the 26S proteasome, for proteasomal degradation. Cul1 is deneddylated by COP9 signalosome.
The potential function of APP in regulating AppBp1 is first examined in studies using molecular and cellular biology techniques. APP’s binding site in AppBp1 includes amino acids 145-251 (Figure 1B) [28]. This AppBp1 fragment was initially discovered by screening a brain cDNA library with GST-C100 (GST fused to APP C-terminal 100 amino acids) as the bait for APP-binding proteins [28]. Further investigations by yeast two-hybrid and co-immunoprecipitation demonstrate that this APP-interacting fragment does not bind Uba3, which does bind AppBp1’s C-terminal 443-534 amino acids [33]. These data suggest that a complex consisting of APP, AppBp1 and Uba3 may assemble in cells. Subsequent structural analyses suggest that AppBp1 (145-251) is essential in the activity of NAE because this region overlaps with the active site that mediates adenylation and also the interaction site between nedd8 and AppBp1. In the structural analyses, NAE is superimposed with MoeB (an ancient form of E1 in bacteria), which shows that the adenylation domain comprises AppBp1’s residues 6–168 and 486–534, and Uba3’s residues 12–210 and 290–347 [43,44]. In addition, AppBp1’s residues 178-280 form a portion of the catalytic cysteine domain with a charged surface that contacts nedd8’s acidic face [45]. These observations strongly suggest that APP plays a role in regulating adenylation and/or nedd8 activation by NAE (Figure 2). Therefore, it is very important to understand the role of APP in the AppBp1 pathway.
AD pathogenesis suggests a gradual loss of APP’s function in cell cycle control
Cell Cycle Markers (CCMs) are often ectopically expressed in AD brain neurons (see review [46]). Table 1 lists examples of the CCMs that are regulated by the AppBp1 pathway and also increased or activated in AD brains. Another CCM Ki67, not expressed in G0 cells, is also significantly increased in AD brain neurons and often co-localizes with neurofibrillary tangles [47,48]. Besides cell cycle proteins, DNA replication in post mitotic neurons is another major CCM. A higher percentage of AD hippocampal neurons enter the S-phase and undergo full or partial DNA re-replication [49,50]. Furthermore, AD neurons may proceed to nuclear division [51]. Such cell cycle events may compromise neuronal function and survival in AD brains [46]. Abnormal CCMs are not limited to neurons. Compared to the controls, AD patient-derived fibroblasts have a two-fold increase in trisomy 21, a mitotic defect due to unequal chromosome segregation [52]. In addition, AD patient-derived lymphocytes are impaired in G1/S checkpoint because they do not respond to cell cycle arrest agent [53]. These pathological changes suggest that the mechanism that prevents ectopic CCM expression is impaired or lost in AD.
What is the protective mechanism that prevents neurons from de-differentiation while they actively respond to all kinds of stimuli? Due to the interaction between APP and AppBp1, the AppBp1 pathway is a good candidate molecular pathway for neuronal survival and function. In one set of studies, APP was overexpressed in primary neurons via the herpes simplex virus vector, which is a valuable model system for investigating potential early triggering events in the development of AD [54]. Overexpression of AppBp1 in primary neurons induces apoptosis through the neddylation pathway [33]. Similarly, overexpression of APP in primary neurons also induces apoptosis by increasing AppBp1 levels [29,55]. In both cases, elevation of AppBp1 levels may increase its insolubility, which forms a potential negative feedback loop that inhibits AppBp1’s activity, causing neuronal death.
Another strategy to determine whether APP regulates AppBp1’s activity is to assay whether APP affects the levels of the substrates downregulated by the AppBp1 pathway. For this purpose, it was first examined whether APP overexpression in primary neurons affected β-catenin, a target of Cullin-1 ubiquitin ligase [56] and it accumulates in Uba3-deficient cells [57] (Figure 2). In primary neurons, a tripling of APP expression via the viral vector leads to a reduction of β-catenin to approximately half of that in the control. Conversely, suppression of the endogenous APP by shRNAs results in a significant increase of total β-catenin compared to the control. The effect of APP on β -catenin was then determined in vivo [56,58,59]. APP knockout neurons have much higher levels of β-catenin in cell bodies and processes than in wild type cells. When β-catenin is stabilized, it is known to translocate to the nucleus where it activates transcription of genes such as cyclin D1. Indeed, cyclin D1 protein levels are also dramatically increased in APP knockout granule cells. These data suggest that APP is an essential component of AppBp1 pathway.
Unlike in primary neuronal cultures where protein expression can be significantly increased, which may mimic an early event in the development of AD, APP levels do not necessarily increase in postmortem brains. Some reports show no overall elevation of APP protein or mRNA in AD brain homogenates [60,61] and others have even reported a decrease [62,63]. One recent study shows that in cognitively intact adult brains, APP accumulates with age, but in AD brains, the protein is not elevated in neurons adjacent to mature plaques [64]. This study also shows that neurons adjacent to mature plaques have dramatically lower levels of APP than those remote from such plaques [64]. APP deficiency can cause age-related cognitive deficits and impaired long-term potentiation in mouse models in vivo [65]. Furthermore, a moderate elevation of APP is neuroprotective in vivo [66]. Similarly, a physiological level of AppBp1 is likely beneficial since it is expressed in some hippocampal neurons in vivo [28,37]. In addition, AppBp1 inhibits amyloid genesis since its suppression by shRNAs results in a dramatic increase of Aβ42 [30].
If the activation of the AppBp1 pathway is a physiological function of APP, the lack of APP in neurons near the plaques in late stage AD may result in a functional deficit of AppBp1. Indeed, pathological changes such as elevation of CCMs observed in postmortem AD brains suggest that the AppBp1 pathway does not function properly (Table 1). AppBp1 activates neddylation of Cullins and neddylated Cullins normally reside in the nucleus [67]. However, in AD hippocampal neurons, almost all nedd8 is present in the cytoplasm, different from the nuclear localization of nedd8 in control neurons [29]. The cytoplasmic localization of nedd8 in AD neurons also suggests that AppBp1 fails to function, which in turn prevents neddylation and activation of Cullin ubiquitin ligases, leading to the accumulation of CCMs in post mitotic neurons. Another pathological change is that more AppBp1 became Triton-insoluble in AD brains [29], which does not occur during normal aging [30]. Similar to the inactivation of AppBp1 in ts41 cells by the non-permissive temperature, cortical precursors from APP-deficient E15 cerebral cortex have defect in crossing G2-M phases [68], further suggesting that APP plays a role in cell cycle transit controlled by AppBp1. Together, these data suggest that the development of AD involves a gradual loss of APP’s function in the AppBp1 pathway , which may be caused by negative feedbacks that warrants further investigation.
![]()
NAE Targets
Function in Cell Cycle
Cullin Ubiquitin Ligase involved
Protein changes in AD brains
Cyclin B
Essential in entry into mitosis [93]
Cul1/ SCF [94]
Increased [95-99]
Cyclin D
Promotes G1-S transition [100]
Cul1/SCF(Fbxw8) [101] Cul1/SCF(Fbx4)[102]
Increased [96,99]
Cyclin E
Endorepilcation in placenta trophoblast giant cells [103]
Cul3 [104]
Cul1/SCF (Skp2) [105-107]
Cul1/SCF(Fbw7) [108]
Increased [97]
Cdc25a
Promote G2-M transistion [109]
Cul1/SCF(�-TrCP) [110]
Activated [111]
p27(kip1)
Nuclear p27 inhibit G1 Phase progression [112,113] Cytoplasmic p27 negatively regulates migration [114,115]
Cul1/SCF [105,116] Cul4 [117]
Increased [118]
Table 1: Examples of the cell cycle proteins degraded by NAE-activated Cullin ubiquitin ligases.
APP in DNA replication and genome integrity
Besides AppBp1, APP also interacts with UV-damaged DNAbinding protein 1 (DDB1) [69], which suggests a role of APP in DNA replication control. DDB1 is the common adaptor subunit in Cul4A and Cul4B (Cul4) ubiquitin ligases [70-73]. DDB1 connects Cul4 with the variable substrate receptor subunit called DDB1-Cul4-Associated Factor (DCAF) [74]. Cul4 ubiquitin ligases are responsible for DNA replication-dependent destruction of Cdt1 in the S-phase of the cell cycle [75-77] and in DNA damage-induced repair synthesis [78,79]. Degradation of Cdt1 in the S-phase prevents DNA rereplication [80] whereas Cdt1 overexpression causes extensive numerical and structural chromosomal aberrations [81]. It has been found that ubiquitination of Cdt1 during the S-phase is coupled to the process of DNA replication [80]. Upon DNA damage, ubiquitination of Cdt1 also localizes to chromatin together with Cul4 [77,82,83]. Together, the evidence suggests that in post mitotic neurons, APP protects genome integrity through DDB1-Cul4 ligase-mediated Cdt1 degradation, but this potential function is gradually lost during disease progression. In fact, the accumulation of unrepaired DNA damage has long been postulated as the cause of neurodegeneration [84-86].
As outlined above, DNA synthesis is coupled to the quality control of genome integrity involving the DNA replication licensing factor Cdt1. Human cells treated with the small molecule inhibitor of NAE, MLN4924, undergo continuous DNA synthesis and have a significant rise in Cdt1 levels among other CCMs [87]. In neurons where overexpression of wild type APP causes DNA synthesis although it has less effect than familial AD APP mutants [29,55], APP overexpression also downregulates Cdt1 levels (Chen’s unpublished observation). These data suggest that a major function of APP in neurons is downregulating Cdt1 when it assembles with AppBp1 and DDB1. Increases in DNA replication in post mitotic neurons from AD brains [46,88] probably represent early events since overexpressing APP or AppBp1 in primary neurons induce DNA syntheses before apoptosis [29]. Together, the data suggest that APP plays a key role in downregulating Cdt1 among others through the AppBp1-activated neddylation pathway, which is likely crucial for DNA damageinduced repair synthesis, which is crucial for the genome integrity of long-lived neurons.
Conclusion
In this review, we analyze the function of APP and hypothesize that APP plays a key role in AppBp1-activated neddylation pathway. In this role, APP protects genome integrity in DNA-damage response and promotes cell cycle progression through the S-M checkpoint. Based on this new hypothesis, the decline of APP’s function is the driving force for the development of AD, resulting in the accumulation of CCMs and neurodegeneration. Repair synthesis in mature neurons is essential for neuronal function if DNA damage can be caused by physiological brain activities [89]. According to the new hypothesis , neuronal vulnerability lies in the inability of efficient repair synthesis due to the dysfunction of APP. APP dysfunction may also impair stem cell differentiation which depends on prior DNA synthesis [90,91]. Although it has not been tested, the function of APP in the AppBp1 pathway may involve APP cleavage into monomeric Aβ peptides since these peptides promote DNA synthesis-linked neural stem cell differentiation [90]. The function of APP may also involve neddylation of APP in the C-terminus [92]. In order to gain a greater understanding of APP’s function as proposed, many questions remain to be answered. What factors cause the gradual decrease of APP’s activity during the development of AD? How does APP activate AppBp1? Further confirmation of the molecular pathway from APP may provide novel drug targets and new therapeutic strategies for AD treatment and prevention.
Acknowledgment
This work was supported by the grant from National Institute of Health/National Institute on Aging (NIH/NIA RO1 AG034980 to Y. C.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services.
References
- Goate A, Hardy J. Twenty years of Alzheimer's disease-causing mutations. J Neurochem. 2012; 120: 3-8.
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2.
- Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 2007; 78: 1158-1159.
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006; 38: 24-26.
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006; 129: 2977-2983.
- Griffin WS, Sheng JG, McKenzie JE, Royston MC, Gentleman SM, Brumback RA, et al. Life-long overexpression of S100beta in Down's syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998; 19: 401-405.
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987; 325: 733-736.
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987; 235: 877-880.
- Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A. 1987; 84: 4190-4194.
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987; 235: 880-884.
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286: 735-741.
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999; 402: 537-540.
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, et al. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999; 402: 533-537.
- Koo EH, Kopan R. Potential role of presenilin-regulated signaling pathways in sporadic neurodegeneration. Nat Med. 2004; 10 Suppl: S26-33.
- Sambamurti K, Suram A, Venugopal C, Prakasam A, Zhou Y, Lahiri DK, et al. A partial failure of membrane protein turnover may cause Alzheimer's disease: a new hypothesis. Curr Alzheimer Res. 2006; 3: 81-90.
- De Strooper B. Aph-, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003; 38: 9-12.
- Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, et al. A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry. 2002; 41: 2825-2835.
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001; 2: 835-841.
- Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem. 2001; 276: 35235-35238.
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, et al. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990; 248: 1122-1124.
- Wang R, Meschia JF, Cotter RJ, Sisodia SS. Secretion of the beta/A4 amyloid precursor protein. Identification of a cleavage site in cultured mammalian cells. J Biol Chem. 1991; 266: 16960-16964.
- Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011; 6: 27.
- Wolfe MS, Guénette SY. APP at a glance. J Cell Sci. 2007; 120: 3157-3161.
- Guo Q, Wang Z, Li H, Wiese M, Zheng H. APP physiological and pathophysiological functions: insights from animal models. Cell Res. 2012; 22: 78-89.
- Chasseigneaux S, Allinquant B. Functions of Aβ, sAPPa and sAPPβ: similarities and differences. J Neurochem. 2012; 120 Suppl 1: 99-108.
- Lazarov O, Demars MP. All in the Family: How the APPs Regulate Neurogenesis. Front Neurosci. 2012; 6: 81.
- Müller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer's disease. Prog Neurobiol. 2008; 85: 393-406.
- Chow N, Korenberg JR, Chen XN, Neve RL. APP-BP, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem. 1996; 271: 11339-11346.
- Chen Y, Liu W, McPhie DL, Hassinger L, Neve RL. APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer's disease brain. J Cell Biol. 2003; 163: 27-33.
- Chen Y, Bodles AM, McPhie DL, Neve RL, Mrak RE, Griffin WS. APP-BP1 inhibits Abeta42 levels by interacting with Presenilin-1. Mol Neurodegener. 2007; 2: 3.
- Cernac A, Lincoln C, Lammer D, Estelle M. The SAR1 gene of Arabidopsis acts downstream of the AXR1 gene in auxin response. Development. 1997; 124: 1583-1591.
- Handeli S, Weintraub H. The ts41 mutation in Chinese hamster cells leads to successive S phases in the absence of intervening G2, M, and G1. Cell. 1992; 71: 599-611.
- Chen Y, McPhie DL, Hirschberg J, Neve RL. The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem. 2000; 275: 8929-8935.
- Hatfield PM, Vierstra RD. Multiple forms of ubiquitin-activating enzyme E1 from wheat. Identification of an essential cysteine by in vitro mutagenesis. J Biol Chem. 1992; 267: 14799-14803.
- Osaka F, Kawasaki H, Aida N, Saeki M, Chiba T, Kawashima S, et al. A new NEDD8-ligating system for cullin-4A. Genes Dev. 1998; 12: 2263-2268.
- Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999; 274: 12036-12042.
- Chen Y, Neve RL, Liu H. Neddylation dysfunction in Alzheimer's disease. J Cell Mol Med. 2012; 16: 2583-2591.
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009; 10: 319-331.
- Kurz T, Pintard L, Willis JH, Hamill DR, Gönczy P, Peter M, et al. Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science. 2002; 295: 1294-1298.
- Haas AL. Structural insights into early events in the conjugation of ubiquitin and ubiquitin-like proteins. Mol Cell. 2007; 27: 174-175.
- Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010; 1: 708-716.
- Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009; 15: 3912-3916.
- Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature. 2003; 422: 330-334.
- Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature. 2001; 414: 325-329.
- Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL Jr, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003; 12: 1427-1437.
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004; 24: 9232-9239.
- Smith TW, Lippa CF. Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995; 54: 297-303.
- Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol. 1997; 93: 294-300.
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001; 21: 2661-2668.
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci. 2007; 27: 6859-6867.
- Zhu X, Siedlak SL, Wang Y, Perry G, Castellani RJ, Cohen ML, et al. Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol. 2008; 34: 457-465.
- Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis. 1999; 6: 167-179.
- Song M, Kwon YA, Lee Y, Kim H, Yun JH, Kim S, et al. G1/S cell cycle checkpoint defect in lymphocytes from patients with Alzheimer's disease. Psychiatry Investig. 2012; 9: 413-417.
- Neve RL, McPhie DL, Chen Y. Alzheimer's disease: a dysfunction of the amyloid precursor protein(1). Brain Res. 2000; 886: 54-66.
- McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, et al. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci. 2003; 23: 6914-6927.
- Chen Y, Bodles AM. Amyloid precursor protein modulates beta-catenin degradation. J Neuroinflammation. 2007; 4: 29.
- Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001; 155: 571-579.
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995; 81: 525-531.
- Mao XR, Moerman-Herzog AM, Chen Y, Barger SW. Unique aspects of transcriptional regulation in neurons--nuances in NFkappaB and Sp1-related factors. J Neuroinflammation. 2009; 6: 16.
- Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, et al. Relative increase in Alzheimer's disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem. 1998; 273: 5013-5019.
- Panegyres PK, Zafiris-Toufexis K, Kakulas BA. Amyloid precursor protein gene isoforms in Alzheimer's disease and other neurodegenerative disorders. J Neurol Sci. 2000; 173: 81-92.
- Harrison PJ, Barton AJ, Procter AW, Bowen DM, Pearson RC. The effects of Alzheimer's disease, other dementias, and premortem course on beta-amyloid precursor protein messenger RNA in frontal cortex. J Neurochem. 1994; 62: 635-644.
- Choi BH, Kim RC, Vaughan PJ, Lau A, Van Nostrand WE, Cotman CW, et al. Decreases in protease nexins in Alzheimer's disease brain. Neurobiol Aging. 1995; 16: 557-562.
- Barger SW, DeWall KM, Liu L, Mrak RE, Griffin WS. Relationships between expression of apolipoprotein E and beta-amyloid precursor protein are altered in proximity to Alzheimer beta-amyloid plaques: potential explanations from cell culture studies. J Neuropathol Exp Neurol. 2008; 67: 773-783.
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience. 1999; 90: 1-13.
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, et al. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997; 78: 135-146.
- Liu HC, Enikolopov G, Chen Y. Cul4B regulates neural progenitor cell growth. BMC Neurosci. 2012; 13: 112.
- López-Sánchez N, Müller U, Frade JM. Lengthening of G2/mitosis in cortical precursors from mice lacking beta-amyloid precursor protein. Neuroscience. 2005; 130: 51-60.
- Watanabe T, Sukegawa J, Sukegawa I, Tomita S, Iijima K, Oguchi S, et al. A 127-kDa protein (UV-DDB) binds to the cytoplasmic domain of the Alzheimer's amyloid precursor protein. J Neurochem. 1999; 72: 549-556.
- McCall CM, Hu J, Xiong Y. Recruiting substrates to cullin 4-dependent ubiquitin ligases by DDB1. Cell Cycle. 2005; 4: 27-29.
- He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006; 20: 2949-2954.
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003; 113: 357-367.
- Shiyanov P, Nag A, Raychaudhuri P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J Biol Chem. 1999; 274: 35309-35312.
- Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell. 2007; 26: 775-780.
- Li JM, Jin J. CRL Ubiquitin Ligases and DNA Damage Response. Front Oncol. 2012; 2: 29.
- Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 2003; 423: 885-889.
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006; 23: 709-721.
- Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003; 5: 1008-1015.
- Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol. 2004; 6: 1003-1009.
- Arias EE, Walter JC. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005; 19: 114-126.
- Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, et al. Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene. 2005; 24: 8176-8186.
- Ishii T, Shiomi Y, Takami T, Murakami Y, Ohnishi N, Nishitani H. Proliferating cell nuclear antigen-dependent rapid recruitment of Cdt1 and CRL4Cdt2 at DNA-damaged sites after UV irradiation in HeLa cells. J Biol Chem. 2010; 285: 41993-42000.
- Roukos V, Kinkhabwala A, Colombelli J, Kotsantis P, Taraviras S, Nishitani H, et al. Dynamic recruitment of licensing factor Cdt1 to sites of DNA damage. J Cell Sci. 2011; 124: 422-434.
- Robison SH, Bradley WG. DNA damage and chronic neuronal degenerations. J Neurol Sci. 1984; 64: 11-20.
- Jones SK, Nee LE, Sweet L, Polinsky RJ, Bartlett JD, Bradley WG, et al. Decreased DNA repair in familial Alzheimer's disease. Mutat Res. 1989; 219: 247-255.
- Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol. 1996; 6: 493-506.
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009; 458: 732-736.
- Arendt T. Cell cycle activation and aneuploid neurons in Alzheimer's disease. Mol Neurobiol. 2012; 46: 125-135.
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, et al. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci. 2013; 16: 613-621.
- McConnell SK, Kaznowski CE. Cell cycle dependence of laminar determination in developing neocortex. Science. 1991; 254: 282-285.
- Lee MR, Lee D, Shin SK, Kim YH, Choi CY. Inhibition of APP intracellular domain (AICD) transcriptional activity via covalent conjugation with Nedd8. Biochem Biophys Res Commun. 2008; 366: 976-981.
- Bassermann F, Peschel C, Duyster J. Mitotic entry: a matter of oscillating destruction. Cell Cycle. 2005; 4: 1515-1517.
- Bassermann F, von Klitzing C, Münch S, Bai RY, Kawaguchi H, Morris SW, et al. NIPA defines an SCF-type mammalian E3 ligase that regulates mitotic entry. Cell. 2005; 122: 45-57.
- Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol. 1997; 94: 6-15.
- Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998; 18: 2801-2807.
- Smith MZ, Nagy Z, Esiri MM. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci Lett. 1999; 271: 45-48.
- Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000; 21: 815-828.
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003; 23: 2557-2563.
- Tashiro E, Tsuchiya A, Imoto M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007; 98: 629-635.
- Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS One. 2006; 1: e128.
- Barbash O, Lin DI, Diehl JA. SCF Fbx4/alphaB-crystallin cyclin D1 ubiquitin ligase: a license to destroy. Cell Div. 2007; 2: 2.
- Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, et al. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J. 2003; 22: 4794-4803.
- Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999; 13: 2375-2387.
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000; 19: 2069-2081.
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001; 413: 316-322.
- Yeh KH, Kondo T, Zheng J, Tsvetkov LM, Blair J, Zhang H. The F-box protein SKP2 binds to the phosphorylated threonine 380 in cyclin E and regulates ubiquitin-dependent degradation of cyclin E. Biochem Biophys Res Commun. 2001; 281: 884-890.
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001; 294: 173-177.
- Li M, Yin S, Yuan J, Wei L, Ai JS, Hou Y, et al. Cdc25A promotes G2/M transition in oocytes. Cell Cycle. 2008; 7: 1301-1302.
- Kanemori Y, Uto K, Sagata N. Beta-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc Natl Acad Sci U S A. 2005; 102: 6279-6284.
- Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer's disease. Am J Pathol. 2000; 157: 1983-1990.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13: 1501-1512.
- Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001; 264: 148-168.
- Nguyen L, Besson A, Roberts JM, Guillemot F. Coupling cell cycle exit, neuronal differentiation and migration in cortical neurogenesis. Cell Cycle. 2006; 5: 2314-2318.
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004; 18: 862-876.
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999; 9: 661-664.
- Miranda-Carboni GA, Krum SA, Yee K, Nava M, Deng QE, Pervin S, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008; 22: 3121-3134.
- Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, et al. Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell. 2003; 2: 105-110.