
Review Article
Austin Alzheimers J Parkinsons Dis. 2014;1(3): 7.
Protein Misfolding and Accumulation as Root Cause in Neurodegeneration
Lonati E1, Sala G2and Bulbarelli A1*
1Department of Health Sciences, Milan Center for Neuroscience University of Milano-Bicocca, Italy
2Department of Surgery and Translational Medicine,Milan Center for Neuroscience University of Milano-Bicocca, Italy
*Corresponding author: Bulbarelli A, Department of Health Sciences Milan Center for Neuroscience University of Milano-Bicocca via Cadore 48, 20900 Monza, Italy.
Received: November 01, 2014; Accepted: November 25, 2014; Published: November 26, 2014
Abstract
The current concept that accumulation and aggregation of misfolded proteins could represent a basic requirement for the neurodegenerative processes, has raised the attention to the efficiency of cell clearance machinery in influencing neuronal protein homeostasis. Indeed, although multifactorial etiology of Alzheimer’s disease (AD) and Parkinson disease (PD) progression, molecular events activated by environmental conditions and epigenetic mechanisms are strongly associated to oxidative stress and inflammatory damage that in turn seems to find a common origin in the anomalous accumulation of misfolded proteins.
Amyloid-β and tau for AD and alpha-synuclein for PD have been proposed as the central and most specific factors implied in the pathogenesis of these syndromes, which, as a consequence, have been classified as a proteinopathies.
Aggregated structures result inappropriate for proteasomal degradation and justify recent studies highlighting the importance of autophagy, a lysosomal degradation pathway, in misfolded proteins degradation in neurons.
Therefore, the poor efficiency of autophagy might be a primum movens in the physiopathology of neurodegenerative diseases. When other pathogenic mechanisms arise, they determine a compensatory induction of autophagy pathways; if this activation is not appropriate to reestablish neuronal homeostasis and prevent accumulation of neurotoxic products, the neurodegenerative process develops.
According to a recent theory regarding cell aging, a major responsible for the death of neurons, would be the neurotoxic effect of aberrant proteins and mitochondria, accumulating within senescent cells as a consequence of an agerelated progressive dysfunction of different catabolic pathways.
Keywords: Alzheimer’s disease; Parkinson’s disease; Amyloid-beta; tau; Alpha-synuclein; Autophagy
Abbreviations
UPS: Ubiquitin/proteasome System; AD: Alzheimer’s disease; PrP: Prion Protein; Aβ: Amyloid beta; PD: Parkinson disease; ALP: Autophagy-Lysosome Pathway; CMA: Chaperone-Mediated Autophagy; Atg: Autophagy Related Genes; hsc70: Heat Shock Cognate Protein; lamp2A: Lysosomal-Associated Membrane Protein 2A; NFTs: Neurofibrillary Tangles; APP: Amyloid Precursor Protein; BACE1: β-secretase; ER: Endoplasmic Reticulum; TGN: Trans-Golgi Network; ROS: Reactive Oxygen Species; (PrP(C)): Cellular Prion Protein, MAP: Microtubules Associated Protein; MTs: Microtubules; PHFs: Paired Helical Filaments; Avs: Autophagosomes; MVBs: Multivesicular Bodies
Introduction
Neurodegeneration referred to neuronal loss is strongly associated with abnormal aggregation of proteins in extra- or intracellular space [1], an event that characterizes the proteinopathies. Pathological protein misfolding and aggregation or aggregation-associated structures (inclusions) have come to be regarded as a deleterious process linked to dysfunction of different neuronal populations and synaptic loss [2,3] that eventually result in cognitive decline/dementia [4] or motor symptom onset [5].
Indeed, among differentiated cell types, neurons are unique in that, because of their extreme polarization, size and post-mitotic nature may be particularly sensitive to the accumulation of aggregated or damaged cytosolic compounds. Hence, protein turnover mechanism alteration, including Ubiquitin-Proteasome System (UPS) and Autophagy-Lysosome Pathway (ALP), becomes a strong detrimental event accompanied by abnormal phosphorylation, ubiquitination, covalent crosslinking and activation of autolytic proteases [6].
The anomalous turnover of aggregation-prone proteins such as alpha-synuclein, Prion Protein (PrP), amyloid-beta (Aβ) peptide, and tau is the basis of 95% neurodegenerative diseases [6]. Alzheime’’s disease (AD) is characterized by the accumulation of Aβ and tau into plaques and tangles, respectively, while Parkinson’s disease (PD) is broadly characterized by the accumulation of alpha-synuclein into Lewy bodies [7]. Oxidative stress, an important pathogenic mechanism in AD [8] and PD [9] contributes to protein misfolding through a double mechanism: oxidative species directly generate post-translational modifications in protein residues, enhancing the tendency of the protein to aggregate; furthermore, high levels of free radicals alter proteins regulating/mediating UPS and ALP and produces ATP depletion through damage of mitochondrial membranes. These effects result in impairment the entire catabolic system of pathogenic proteins such as Aβ, tau and alpha-synuclein. Oxidative stress also represents an important factor both favoring and mediating glutamate-induced excitotoxicity occurring in AD and PD [10]. The ROS-induced lipid peroxidation of presynaptic membranes impairs the function of transporters involved in the maintenance of calcium homeostasis, with consequent glutamate release into the synaptic cleft, and of glutamate transporters, leading to an increase of extracellular glutamate, which can bind postsynaptic receptors and mediate excitotoxicity. Although pathogenic proteins associated to AD and PD are different, their ‘intrinsically disordered’ structure may contribute to clearance mechanism disruption, leading to the formation of microaggregates, that, in turn, can be combined and deposited into larger aggregates of various size and structure [11].
The recently emerged concept that the accumulation and aggregation of misfolded proteins could represent a basic requirement for the neurodegenerative process has raised the challenge of establishing the pathogenic role of the biological systems influencing neuronal protein homeostasis [12].
Intracellular catabolic systems
The Ubiquitin-Proteasome System (UPS) and the Autophagy- Lysosome Pathway (ALP) are the two major systems responsible for intracellular protein degradation.
The UPS is a major cellular mechanism required for the targeted degradation of most short-lived proteins, thus ensuring protein quality control in both the cytoplasm and the nucleus. The UPS is a ~ 2.5 MDa holoenzyme complex that comprises the 28-subunit core particle (20S subunit), and the 19-subunit regulatory particle (19S subunit). UPS recognizes, unfolds, and degrades polyubiquitinated substrates into small peptides of 3-24 amino acids. For degradation, UPS substrates are covalently conjugated with ubiquitin, a highly conserved 76-residue protein, through a complex enzymatic cascade involving E1, E2, and E3 enzymes [13]. E1 ubiquitin-activating enzymes generate a high-energy thioester bond between the C-terminus of ubiquitin and cysteine residues in E1. E2 ubiquitinconjugating enzymes transfer the activated ubiquitin to the E3- substrate complex. The abundance and specificity of the currently identified E3 ligases suggest that these enzymes determine the substrate selectivity of the UPS.
The ALP is a finely regulated and conserved cellular “selfeating” process that involves sequestration and delivery of cytosolic components to the lysosomes for degradation and recycling. Its function allows the clearance of substrates characterized by alterations limiting their physiologic function or responsible for a cytotoxic effect. This degradative process exerts a cytoprotective role that is probably dependent on the clearance of toxic intracellular structures and the catabolism of substrates in order to obtain energy during starvation. Anyway, in particular situations autophagy seems to mediate a specific pathway of programmed cell death; this function requires a strong activation of autophagy and until now, in vivo, has been identified only during involutional physiologic processes in embryonic tissues [14].
Three types of autophagy, depending on their respective sequestration and delivery mechanism, are known: microautophagy, macroautophagy and Chaperone-Mediated Autophagy (CMA) [15]. Microautophagy is a constitutive, non-selective process consisting on endocytosis of small amounts of cytoplasm into lysosomes through invagination of lysosomal membrane. Macroautophagy and CMA are inducible processes. Macroautophagy, responsible for the removal of misfolded proteins and aberrant organelles which are unsuitable for degradation via UPS, proceeds through various steps, each requiring the presence of specific Autophagy Related Genes (Atg). Macroautophagy starts with sequestration of a region of cytoplasm containing proteins and organelles designed for degradation within a double-membrane vacuole called autophagosome. Once formed, the autophagic vacuole undergoes a process of maturation, which is essential for the subsequent fusion with lysosome and the degradation of substrates.
CMA is a selective device for degradation of aberrant proteins, containing the consensus peptide sequence KFERQ, which are directly transported into the lysosomal lumen by a translocation system constituted by specific carrier proteins. CMA process requires the presence of two main proteins: cytosolic and lysosomal heat shock cognate protein 70 (hsc70) and lysosomal-associated membrane protein 2A (lamp2A). Cytosolic hsc70 binds the KFERQ sequence of substrate proteins and carries them to the lysosomal membrane, where lamp2A, after interaction with cytosolic hsc70, multimerizes and forms a translocation complex with lysosomal hsc70, thus mediating the transport of the substrate protein into the lysosomal lumen. The binding of the substrate protein to lamp2A represents the limiting step of CMA.
The current knowledge indicates that UPS and ALP are not parallel and independent proteolytic pathways but rather compensatory mechanisms cooperating in protein quality control. As a matter of fact, a functional interaction among them has been demonstrated. In particular, macroautophagy is able to modulate its own activity depending on the efficiency of the other two pathways. Both proteasome and CMA inhibition determine a cytoprotective activation of macroautophagy.
Alzheimer’s disease
AD is the most common cause of dementia in the elderly population, affecting approximately 7% of people older than 65 years and about 40% of people older than 80 years [16].
AD progression, indeed, leads slowly to loss of neurons in brain regions such as hippocampus, amygdala and cerebral cortex, destroying memory and cognitive functions.
Although its multifactorial etiology, molecular events activated by environmental conditions or epigenetic mechanisms converge altogether to the increase of hippocampal dystrophic neurites and synapse failure: number of synapses are already reduced in patients with mild cognitive impairment, a preclinical stage of AD, along with a compensatory increase in size of the remaining ones [17]. The described morphology changes are strongly associated to oxidative stress and inflammatory damage that in turn seem to find a shared origin in the anomalous accumulation of misfolded proteins [18].
In AD post-mortem brains, aggregates of nonfunctional protein tau called Neurofibrillary Tangles (NFTs) and cerebral plaques laden of the toxic Aβ peptide have been principally detected. Nonetheless, in the last years, intermediate oligomers and aggregates of Aβ as well as of abnormal tau have been reported as the most cytotoxic forms [18-20].
The imbalance between aggregation and clearance of these forms may easily lead to the following oxidative and inflammatory events underlying AD neurodegeneration [21]. Indeed, several defects in autophagy machinery (i.e. autophagosome formation, lysosomal acidification) has been identified in AD mouse models, suggesting the importance of its activation in preventing amyloidosis and tau aggregation [22].
Amyloid beta and tau: Aβ is a short peptide of about 39-42 amino acids generated by proteolytic cleavage of the membrane-bound Amyloid Precursor Protein (APP), by sequential enzymatic actions of β-secretase (BACE1), producing βAPPs and C99, and the following cleavage by a large multiprotein complex known as γ–secretase (Figure 1A) [23]. APP, synthesized in Endoplasmic Reticulum (ER), completes its maturation in the Trans-Golgi Network (TGN) to be delivered to the plasma membrane [24]. A quote of APP is recycled and internalized in endosome vesicles, where the amyloidogenic processing appears to prevalently occurs [24]. Indeed, in endosomal/ lysosomal vesicles, BACE1 working at the optimal pH is strongly activated.
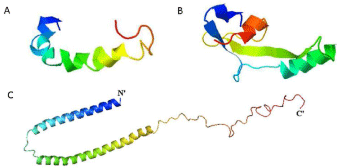
Figure 1: AB42, tau and a-synuclein crystal structures : A) Proposed AB42
structure based on predicted β-strand propensities (Displayed in Jmol) B)
Proposed PHF core structure based on predicted β-strand propensities
(Displayed in Jmol) [121] C) Structure of alpha-synuclein bound to lipid
vesicles. The structure of the full-length human protein was determined by
NMR (PDB ID: 1XQ8).
Moreover, all through APP maturation, a certain amount of Aβ may be produced in ER and retained in an insoluble state [25]. Recognized as a misfolded protein, the peptide may be translocated back to cytosol and targeted for degradation. Since the native conformation of Aβ has little secondary structure, this peptide spontaneously self-aggregates from monomers into oligomers and/ or in fibrils according to the natively unfolded protein model [26]. Indeed, its native random coil rich state is folded to a a-helical rich intermediate and finally to a β–sheet rich amyloid monomer that can self-assemble. Dimers and oligomers formation may depend on a core structure located around the central hydrophobic cluster (residues 17–21) representing a possible anchor point for the molecular recognition of other proteins in analogous conformations [27]. The buildup of intracellular Aβ could contribute to mitochondrial damage leading to electron transport, ATP production, oxygen consumption, mitochondrial membrane potential impairment, and mitochondrial Reactive Oxygen Species (ROS) generation [28]. As well as, the excessive accumulation of A in soluble species can permeate and disrupt cell phospholipid membranes ultimately lead to leakage of ions across the membrane and cause an influx of calcium ions into the cytosol from the surroundings and endoplasmic reticulum [18].
Moreover, Aβ oligomer neurotoxic effect has been associated with the cellular Prion Protein (PrP(C)) recruitment at the plasma membrane, where Aβ-PrpC direct or indirect binding with NMDA form an ectopic signaling platform [29,30].
Tau is a Microtubule Associated Protein (MAP) expressed, in adult central nervous system, in six different isoforms derived by single gene alternative splicing. Each isoform contains a N-terminal ‘projection domain’, rich in proline, and a C-terminal microtubule binding domain constituted of three or four repeats of highly conserved tubulin binding motifs (Figure 1B) [6]. The proline rich region of tau allows its classification as an intrinsically disordered protein, whose structural flexibility is required to perform important role in many cellular processes, although this characteristic favors tau acquirement of a rigid fold and following formation of highly insoluble filaments [31]. Major physiological function of tau is related to its axonal localization, where the protein promotes assembly and stability of Microtubules (MTs) [32], contributes to axonal identity (i.e., neuronal polarization), outgrowth [6], neuronal differentiation, and regulates axonal transport of signalling molecules, trophic factors and organelles [33]. Nevertheless, tau may play other physiological functions associated to its nuclear [34] and plasma membrane [35] localization.
For all these reasons tau protein is finely regulated by posttranslational modifications: phosphorylation, glycosylation, glycation, prolyl-isomerization, cleavage or truncation, nitration, polyamination, ubiquitination, sumoylation and oxidation [36]. In particular, the balance between tau phosphorylation and dephosphorylation global state is the main regulatory mechanism in tau-MT association. Therefore, this equilibrium is strictly modulated by Pin1-mediated prolyl-isomerization on specific Thr231 amino acid, that in turn allows dephosphorylation of other key serine/ threonine residues [37]. Indeed, high phosphorylation levels in specific serine/threonine sites (S262, S293, S324 and S356, Thr231, Ser396 and Ser404) determine tau dissociation from MTs [36,38]. Then, soluble hyperphosphorylated tau, accumulating in the somatodendritic compartment of neurons [39], is more prone to aggregation in oligomers that in turn rearrange into Paired Helical Filaments (PHFs) and finally self-assemble in NFTs.
Following tau hyperphoshorylation, protein aggregates are characterized by several pro aggregation post-translational modifications [36]. Above all, tau truncation in C-terminal has been identified as a key event in the NFTs formation [40]. Caspase-3 proteolytic cleavage of C-terminal at D421 [41,42] leads to the formation of fragments, that interacting each other may also sequester full-length tau [6] contributing to neuronal degeneration. Indeed, accumulation of truncated fragments has been identified in AD brains and correlates with AD progression [43]. Moreover also tau abnormal ubiquitination is a post-phosphorylation modificationincluded in the pro-aggregation group, probably occurring also after protein truncation [36].
Accumulation of misprocessed tau has been associated to dendritic and axonal changes in both transgenic mouse [44] and AD brains [45]. As Aβ oligomers also tau aggresomes contribute to mitochondria dysfunction and abnormal distribution, low ATP, plasma membrane permeability and calcium ions-mediated toxicity [46].
Thus, tau and Aβ interplay influencing their correct folding and/or turnover. It has already been demonstrated that Aβ increase and accumulation may induce tau aggregation promoting caspase- 3-dependent tau cleavage [47] and glycogen synthase kinase 3β activation [48]. On the other hand, the tau regulation of APP axonal transport [49] is impaired by mis-processed tau, leading to Aβ generation and deposition. In addition, Ittner and colleagues demonstrated that tau dendritic localization mediates Aβ toxicity at the post-synaptic compartment [35].
Therefore monomers or soluble oligomers are physiologically removed through the ALP and/or the UPS in order to maintain the equilibrium between rate of aggregation and clearance.
Dysfunction of catabolic pathways in AD: AD progression is characterized and sustained by the UPS and ALP impairment with a progressive collapse of the intracellular proteolysis. Although early dysfunctions of the clearance mechanisms are related to aging, oxidative stress and/or other environmental causes, also both Aβ and tau aggregates directly or indirectly promote the proteasomemediated and the autophagic- mediated clearance damage, accelerating their own accumulation [18].
Elevated levels of Aβ oligomers have been shown to inhibit the 26S subunit of proteasome [50,51], with the consequent A, APP and BACE1 turnover disruption. A major availability of APP and BACE1 results in the amyloidogenic pathway increment and oligomers formation. As well as, inhibition of UPS induced accumulation of Ub-conjugates [52], including abnormal ubiquitinated tau, then sequestered in PHFs and NFTs [53].
Unable to be degraded by UPS system, the ubiquitination of these aggregates may also act as a signal tag in the ALP [11]. The autophagic receptor p62 recognizes Ub-tau aggresomes through the C-terminal ubiquitin associated domain p62 [54,55] targeting them to Autophagosomes (AVs) [11]. However, an increase of p62 positive tau inclusions, probably related to failure of autophagosomal machinery, has been observed in AD brains [56,57]. Accordingly, it has been observed that AVs accumulate in swollen dystrophic neurites in AD mouse model and human brains [24,58,59], concomitantly to a reduction in beclin1 [60]. Beclin1 reduction correlates to an increase in APP and its metabolites and also to a reduction in AV degradation, resulting in LC3-II increase [60]. Moreover, the hybrid `amphisome’ structures derived by the fusion between autophagosomes and late endosomes/Multivesicular Bodies (MVBs) [61,62] represent a suitable milieu for the amyloidogenic pathway. Indeed, co-residence of APP, BACE1 [63] and components of the γ-secretase complex [64] along to the internal pH create the favorable environment for either production or degradation of A [24]. Although A produced in AVs should be delivered and rapidly degraded by cathepsins in lysosomes, autophagy activation in AD results in accumulation of A toxic storage [65], according to the impairment of autophagosome maturation in autolysosome [66]. As well as, tau contributes to the accumulation of AVs filled in ubiquitinated aggregates, since it appears to interfere with acidification of autolysosomes [67]. In fact, caspase-cleaved tau fragment degradation, preferentially degraded by both macroautophagy and CMA [68,69], looks to be a neurotoxic effector of lysosomal dysfunction in AD [70] associated to loss of cathepsin D. In addition, AD-associated FTP-17 mutation in tau gene has been reported to impair CMA function through disruption of lysosomal membrane integrity [69].
However, in the last years it has been elucidated an unconventional autophagy activation that, instead of the classical degradation fate, eventually results in secretion of own protein content through a pathway called exophagy [71,72]. Exophagy is based on the idea that autophagosomes and amphisomes may exchange their contents with endocytic recycling compartments or fuse directly with the plasmamembrane [24,67].
Both A and tau might be released in the extracellular space, in line with other tau secretion pathway, such as exosomal secretion, microvesicle shedding [73,74] and a novel ectosome pathway [75]. Lee and colleagues have demonstrated, in a lamprey ABC tauopathy model, a co-localization of tau and LC3 positive vesicles that appear to be exocytosed [67]. In the case of tau, phosphorylation and cleavage of the protein at D421 [76] favour its secretion. On the other side, a fraction of the secreted A peptide is specifically generated along the autophagy route [24] (Figure 2). It is worth to note that A and tau toxicity might be interneuronal spread, since A monomers or oligomers as well as the misprocessed or aggregated tau secreted might be uptaken by healthy neurons, where spontaneously selfassemble and stimulate further misfolding and protein aggregation [6,77].
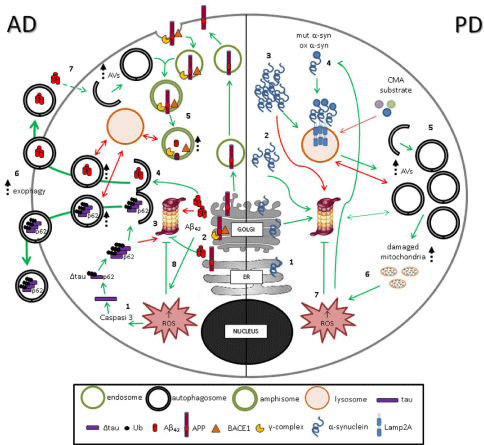
Figure : Impairment of catabolic pathways in AD and PD. AD- In AD cells, Δtau and A42 , produced by caspase3 (1) and BACE1/-complex (2) cleavage
respectively, are aggregated in oligomers and PHFs. Aggregates of A42 and ubquitinated Δtau, which cannot be degraded by UPS (3), enter in macroautophagic
catabolic pathway (4). AVs fuse with endosomes recycled from plasma membrane resulting in amphisomes, suitable milieu for further A42 production (5). The
number of AVs and amphisomes, unable to fuse with lysosomes, increases, and the incomplete macroautophagy process leads to exophagy of vescicles containing
A42 and ubquitinated Δtau aggregates (6). A42, in turn, sustains autophagy activation (7), inhibits proteasome (8), and induces ROS production and damageto
mitochondria, favoring aggregate formation. PD- Soluble (1) and oligomeric (2) a-synuclein is degraded by both UPS and autophagy. Aggegated (3) a-synuclein
cannot be degraded by UPS. Mutant and oxidant-modified forms of a-synuclein (4) bind abnormally to Lamp2A leading to blockage of their own degradation as
well as degradation of other CMA substrates. An induction of macroautophagy occurs, characterized by an increased number of AVs (5) which are unable to fuse
with lysosomes thus leading to an incomplete macroautophagy process. As a result, damaged mitochondria (6) accumulate, with consequent ROS production (7)
responsible for a further UPS inhibition and oxidation of a-synuclein (4) in a vicious loop.
Finally, it has been demonstrated that Aâ itself, inducing ROS generation, may activate autophagy in a sort of feedback loop to promote its own degradation, eventually resulting in further production of toxic peptides [66,78]. Moreover, Nah and colleagues showed that, in response to Aâ42, autophagy activation is also mediated by interaction between PrP and beclin1 in lipid rafts [79].
Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after AD, characterized by progressive loss of dopaminergic neurons in the substantia nigra and leading to impairment of patient’s motor skills, speech and other functions. Classical motor symptoms are usually preceded by non-motor manifestations due to dysfunction within other areas of the central and peripheral nervous system.
At present the molecular mechanisms involved in PD pathogenesis are not fully understood and PD is considered a multifactorial disease resulting from a complex interplay between different genetic and environmental factors [5]. Approximately 15% of PD patients have a family history of this disorder. Familial cases are caused by mutations in known genes (alpha-synuclein, parkin, PINK1, DJ-1, LRRK2, GBA) or by alterations in other not yet identified genes. Mutations or polymorphisms may also play a role in cases that appear to be sporadic, as a background for age-related environmental factors.
Alpha-synuclein has been proposed as the central and most specific factor implied in the pathogenesis of this syndrome, which, as a consequence, has been classified as a proteinopathy.
Alpha-synuclein: Alpha-synuclein is a 144 amino acid protein encoded by the gene SNCA, localized at 4q chromosome (Figure 1C). Three different SNCA transcripts can be detected in neurons. The physiological functions of alpha-synuclein is still not fully understood. It is abundantly expressed in nervous tissues and localizes in the cytoplasm or is associated with lipid membranes. In particular, this protein is mainly localized in the pre-synaptic compartment, where it seems to have a role in regulating neurotransmitter release, vesicle turnover, membrane stability and neuronal plasticity [80]. Alphasynuclein is a typical intrinsically disordered protein which can adopt a number of different conformational states depending on conditions and cofactors. These include the helical membrane-bound form, a partially-folded state that is a key-intermediate in aggregation and fibrillation, various oligomeric species, and fibrillar and amorphous aggregates [81]. The hypothesis of an active involvement of alphasynuclein in the pathogenesis of PD was proposed when this protein was identified as the main component of Lewy bodies, intraneuronal aggregates constituting a constant report in the neuropathology of PD [82]. The key function of alpha-synuclein protein in the pathogenesis of PD was first elucidated by genetic studies: the finding of familial cases of PD genetically linked to missense mutations in the SNCA gene and to genomic triplication of the wild-type gene gave evidence of the potential neurotoxic effect of this protein [83-86]. The deleterious effects of these genetic alterations on dopaminergic neurons have been demonstrated in cellular and animal models, confirming the hypothesis that both qualitative and quantitative alterations of alphasynuclein are able to trigger its toxic effect [87,88].
The toxicity of both mutant and wild-type alpha-synuclein seems to require the acquisition of a misfolded conformation which prevents alpha-synuclein degradation and favors its fibrillation, firstly into protofibrillar oligomeric species and then to fibrillar aggregates [89]. Insoluble aggregates seem do not have an intrinsic toxic function but rather represent a protective phenomenon favoring the removal of soluble oligomers responsible for the neurotoxic effect of the protein [90]. Based on the demonstration of the existence of an exosome-mediated inter-neuronal transport of alpha-synuclein [88], a prion-like mechanism favoring the spreading of alpha-synuclein neurotoxicity has been indicated [91]. As well as in familial cases, even in sporadic PD the toxic gain of function of alpha-synuclein could derive from the intraneuronal accumulation of the protein or from biochemical modifications enhancing the propensity of the protein to aggregate [92,93]. Oxidative stress and production of highly reactive aldehydes, both depending on high levels of dopamine, can lead to alpha-synuclein post-translational modifications consisting in oxidation and nitration of specific amino acid residues, partially explaining the high tendency of this protein to acquire a misfolded conformation in dopaminergic neurons. These alterations in alpha-synuclein have been detected in nigral neurons and lymphomonocytes from PD patients [94,95] and animal models of disease [96,97].
Dysfunction of catabolic pathways in PD: The finding that Lewy bodies are ubiquitin-positive aggregates has suggested that a dysfunction in proteasome might contribute to the accumulation and aggregation of alpha-synuclein and other neurotoxic proteins. The first confirmation of this hypothesis derived from the identification of hereditary forms of PD linked to mutations in two genes within the UPS: parkin, an ubiquitin E3 ligase, and UCH-L1, an enzyme that cleaves peptide-ubiquitin bonds and recycles ubiquitin monomers [98,99].
The finding of structural and functional alterations in the 20S proteasome subunit in the substantia nigra of patients with sporadic disease has confirmed the existence of UPS dysfunction in PD [100]. The pathogenic relevance of proteasome impairment has been reinforced by the observation that administration of proteasomal inhibitors to animals produces the neuropathological and motor manifestations of PD, including selective nigral cell loss, Lewybodies- like inclusions and typical clinical signs [101].
Aging, associated with a physiologic decrease of proteasome efficiency and ubiquitination activity, oxidative species and alphasynuclein protofibrils, exerting a deleterious effects on proteasome subunits, are recognized to be responsible for UPS dysfunction. Alphasynuclein oligomers were also demonstrated to inhibit proteasome function through direct interaction with 20S subunit [102], and this effect might favor further accumulation of alpha-synuclein, which in turn may worsen proteasome impairment.
Although an impairment of UPS occurs in PD, it does not seem to exert a remarkable clearance function on alpha-synuclein. More recently, a correlation between the efficiency of the ALP and nigral degeneration has been proposed.
A role for macroautophagy in the development of PD was proposed when intraneuronal accumulation of autophagic vacuoles was detected in postmortem brains of patients [103]. Increasing experimental data indicate that this feature is likely to mirror a dysfunction in maturation and lysosomal clearance of autophagosomes, that physiologically occurs in aged longlived cells as neurons [104,105]. Both pathogenic mutations and over-expression of alpha-synuclein determine an induction of macroautophagy, which is dependent on the inhibition of CMA [106]. Anyway, the impairment of alpha-synuclein degradation seems not to have a major role in mediating the deleterious effect of macroautophagy impairment: the accumulation of other substrates might be more decisive. In this regard, it is important to remind that macroautophagy represents the only mechanism able to mediate the clearance of damaged mitochondria through a process named mitophagy [107]. The intraneuronal accumulation of aberrant mitochondria determines neurotoxic effects linked to the generation of reactive oxygen species and the release of pro-apoptotic mediators. Increasing evidence from transgenic models of disease suggests that a defect in the mitophagy pathway might exert an key pathogenetic role in PD (Figure 2). In fact, genes responsible for hereditary disease are essential components of mitophagy machinery: PINK1 and parkin, two genes linked to recessive forms of PD, encode proteins that work synergistically to ensure the sequestration of aberrant mitochondria within the autophagic vacuole [108]. Even DJ1, a gene linked to autosomal recessive PD, encodes for a protein that activates macroautophagy and favors mitochondrial turnover [109]. Loss of function of PINK1, parkin or DJ1 causes hereditary PD and the death of dopaminergic neurons in cell and animal models: the decreased efficiency of macroautophagy and mitophagy might be responsible for part of this neurotoxic effect.
The demonstration that CMA is the main catabolic pathway for alpha-synuclein [110,111] and that this protein accumulates when CMA is suppressed by lamp2A expression down-regulation indicated an involvement of this pathway in the pathogenesis of PD [112]. Furthermore, both pathogenic mutations and overexpression of alpha-synuclein are known to inhibit this process [106,110,113]. Experiments performed on animal models over-expressing alphasynuclein have revealed that down-regulation of CMA is responsible for part of the alpha-synuclein toxicity in dopaminergic neurons. A reduced turnover of proteins directly involved in the neuronal survival and in the apoptotic machinery may exacerbate the deleterious effect of alpha-synuclein-mediated inhibition of this clearance pathway. Therefore, it is conceivable to assume that other substrates of CMA, including the neuronal survival factor MEF2D, contribute to neuronal death through their accumulation [114]. Besides mutations and multiplications of alpha-synuclein gene, other mutations linked to hereditary PD are responsible for the impairment of CMA. Pathogenic mutation I93M of UCH-L1 gene has been demonstrated to determine the inhibition of CMA and the accumulation of alphasynuclein [115,116] (Figure 2). Recently, the G2019S mutant LRRK2 has been found to interfere with the organization of the CMA translocation complex, thus causing defective CMA [117-118].
The finding of low levels of lamp2A and hsc70 in post-mortem substantia nigra of patients with sporadic disease [112] and the presence of some of these alterations also in patient lymphomonocytes [119] indicate that a reduced activity of CMA is likely to be a pathogenic mechanism even in idiopathic PD.
Conclusion
Collectively, this review points up the pathogenic importance of protein misfolding, accumulation and aggregation in the onset of the two most common neurodegenerative diseases, AD and PD. Hence, the efficiency of intracellular protein clearance machinery appears as a crucial step in determining neuronal susceptibility to protein toxicity. ALP and UPS act as control systems able to rescue neurons from the deleterious mechanisms causing the degenerative process. According to this hypothesis, the progressive neuronal degeneration would start only when the efficiency of these pathways is compromised enough to be harmful and lead to the development of processes exerting a direct toxic function. Therefore, the poor efficiency of ALP and UPS might be a primum movens in the physiopathology of neurodegenerative diseases such as AD and PD (Figure 3). When other pathogenic mechanisms arise, they determine a further induction of autophagy pathways; if this activation is unable to reestablish neuronal homeostasis, neurotoxic products accumulate. Several studies reported a decrease in the activity of both ALP and UPS during aging in every tissue, including neurons [105,120]. Aberrant proteins and damaged mitochondria, accumulating within senescent cells as a consequence of an age-related progressive dysfunction of different catabolic pathways, would be the major responsible for the neuronal death. A deep investigation of the molecular mechanisms governing the cell catabolic machinery is needed in order to enhance the degradation of aggregate-prone proteins and to translate this strategy into effective therapeutic approaches for neurodegenerative diseases.
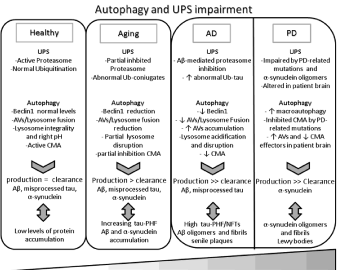
Figure 3: Schematic representation of UPS and autophagy impairment. In healthy neurons, the physiologic catabolic system activity allows to maintain a
balance between production and clearance of A, misprocessed tau and a-synuclein. During aging, UPS and autophagy pathways are partially inhibited leading
to an increase of protein accumulation. In neurodegeneration catabolic systems are impaired by the synergic effect of AD- and PD-related
pathogenic mechanisms and aging, favoring the aggregation of misfolded proteins and toxic peptides in oligomers and fibrils, that, in turn, sustain UPS/
autophagy damage in a vicious loop.
References
- Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. 2013; 126: 365–384.
- Amen T, Kaganovich D. Dynamic droplets: the role of cytoplasmic inclusions in stress, function, and disease. Cell Mol Life Sci. 2014;.
- Vandenberghe R. The relationship between amyloid deposition, neurodegeneration, and cognitive decline in dementia. Curr Neurol Neurosci Rep. 2014; 14: 498.
- Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013; 74: 478-489.
- Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson's disease. Mov Disord. 2011; 26: 1049-1055.
- Gendreau KL, Hall GF. Tangles, Toxicity, and Tau Secretion in AD - New Approaches to a Vexing Problem. Front Neurol. 2013; 4: 160.
- McMillan PJ, Leverenz JB. From model system to clinical medicine: pathophysiologic links of common proteinopathies. Alzheimers Res Ther. 2010; 2: 26.
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997; 23: 134-147.
- Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998; 44: S72-84.
- Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999; 81: 163-221.
- Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010; 584: 2635-2645.
- Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Rossi G, Keller JN, et al. Wild type and mutant amyloid precursor proteins influence downstream effects of proteasome and autophagy inhibition. Biochim Biophys Acta. 2014; 1842: 127-134.
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001; 70: 503-533.
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006; 29: 528-535.
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007; 8: 931-937.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140: 918-934.
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007; 68: 1501-1508.
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010; 362: 329-344.
- Flach K, Hilbrich I, Schiffmann A, Gärtner U, Krüger M, Leonhardt M, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem. 2012; 287: 43223-43233.
- Ow SY, Dunstan DE. A brief overview of amyloids and Alzheimer's disease. Protein Sci. 2014; 23: 1315-1331.
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009; 110: 1129-1134.
- Nassif M, Hetz C. Autophagy impairment: a crossroad between neurodegeneration and tauopathies. BMC Biol. 2012; 10: 78.
- Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004; 5: 499-504.
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007; 120: 4081-4091.
- Li M, Chen L, Lee DH, Yu LC, Zhang Y. The role of intracellular amyloid beta in Alzheimer's disease. Prog Neurobiol. 2007; 83: 131-139.
- Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002; 11: 739-756.
- Flöck D, Colacino S, Colombo G, Di Nola A. Misfolding of the amyloid beta-protein: a molecular dynamics study. Proteins. 2006; 62: 183-192.
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2010; 107: 14164-14169.
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet. 2012; 21: 1138-1144.
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010; 66: 739-754.
- Skrabana R, Skrabanova M, Csokova N, Sevcik J, Novak M. Intrinsically disordered tau protein in Alzheimer's tangles: a coincidence or a rule? Bratisl Lek Listy. 2006; 107: 354-358.
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007; 8: 663-672.
- Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005; 109: 5-13.
- Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011; 286: 4566-4575.
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010; 142: 387-397.
- Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2011; 58: 458-471.
- Bulbarelli A, Lonati E, Cazzaniga E, Gregori M, Masserini M. Pin1 affects Tau phosphorylation in response to Abeta oligomers. Mol Cell Neurosci. 2009; 42: 75-80.
- Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009; 48: 10047-10055.
- Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Börki K, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995; 14: 1304-1313.
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. 2010; 464: 1201-1204.
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004; 114: 121-130.
- Zhao H, Zhao W, Lok K, Wang Z, Yin M. A synergic role of caspase-6 and caspase-3 in Tau truncation at D421 induced by H2O 2. Cell Mol Neurobiol. 2014; 34: 369-378.
- Basurto-Islas G, Luna-Muñoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, García-Sierra F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008; 67: 470-483.
- Spittaels K, Vanden Haute C, VanDorpe J, Bruynseels K, Vandezande K, Laenen I, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 1999; 155: 2153–2165.
- Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol. 1987; 22: 639-643.
- Furukawa Y, Kaneko K, Nukina N. Tau protein assembles into isoform- and disulfide-dependent polymorphic fibrils with distinct structural properties. J Biol Chem. 2011; 286: 27236-27246.
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006; 281: 20315-20325.
- Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010; 223: 322-325.
- Adalbert R, Gilley J, Coleman MP. Abeta, tau and ApoE4 in Alzheimer's disease: the axonal connection. Trends Mol Med. 2007; 13: 135-142.
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008; 29: 1607-1618.
- Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer's disease. Neurol Res. 2014; 36: 276-282.
- Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Rossi G, Buizza L, et al. Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer's disease. Biochim Biophys Acta. 2012; 1822: 1741-1751.
- Riederer IM, Schiffrin M, Kövari E, Bouras C, Riederer BM. Ubiquitination and cysteine nitrosylation during aging and Alzheimer's disease. Brain Res Bull. 2009; 80: 233-241.
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005; 94: 192-203.
- Tung YT, Wang BJ, Hsu WM, Hu MK, Her GM, Huang WP, et al. Presenilin-1 regulates the expression of p62 to govern p62-dependent tau degradation. Mol Neurobiol. 2014; 49: 10-27.
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002; 160: 255-263.
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009; 174: 1241-1251.
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005; 64: 113-122.
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996; 16: 5795-5811.
- Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010; 5: e11102.
- Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002; 17: 897-908.
- Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2.
- Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. The Alzheimer’s ß-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013; 126: 329-352.
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004; 36: 2531-2540.
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005; 171: 87-98.
- Tung YT, Wang BJ, Hu MK, Hsu WM, Lee H, Huang WP, et al. Autophagy: a double-edged sword in Alzheimer's disease. J Biosci. 2012; 37: 157-165.
- Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int J Alzheimers Dis. 2012; 2012: 172837.
- Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010; 285: 21978-21987.
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009; 18: 4153-4170.
- Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, et al. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010; 6: e1001026.
- Abrahamsen H, Stenmark H. Protein secretion: unconventional exit by exophagy. Curr Biol. 2010; 20: R415-418.
- Manjithaya R, Subramani S. Autophagy: a broad role in unconventional protein secretion? Trends Cell Biol. 2011; 21: 67-73.
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012; 287: 3842-3849.
- Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012; 287: 20522-20533.
- Dujardin S, Bégard S, Caillierez R, Lachaud C, Delattre L, Carrier S, et al. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One. 2014; 9: e100760.
- Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One. 2012; 7: e36873.
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009; 284: 12845-12852.
- Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009; 5: 502-510.
- Nah J, Pyo JO, Jung S, Yoo SM, Kam TI, Chang J, et al. BECN1/Beclin 1 is recruited into lipid rafts by prion to activate autophagy in response to amyloid β42. Autophagy. 2013; 9: 2009-2021.
- Lavedan C. The synuclein family. Genome Res. 1998; 8: 871-880.
- Breydo L, Wu JW, Uversky VN. A-synuclein misfolding and Parkinson's disease. Biochim Biophys Acta. 2012; 1822: 261-285.
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997; 388: 839-840.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997; 276: 2045-2047.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003; 302: 841.
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998; 18: 106-108.
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004; 55: 164-173.
- Dawson T, Mandir A, Lee M. Animal models of PD: pieces of the same puzzle? Neuron. 2002; 35: 219-222.
- Vekrellis K, Rideout HJ, Stefanis L. Neurobiology of alpha-synuclein. Mol Neurobiol. 2004; 30: 1-21.
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000; 97: 4897-4902.
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009; 106: 13010-13015.
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010; 33: 317-325.
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. 2001; 21: 8053-8061.
- Hodara R, Norris EH, Giasson BI, Mishizen-Eberz AJ, Lynch DR, Lee VM, et al. Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem. 2004; 279: 47746-4753.
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000; 290: 985-989.
- Prigione A, Piazza F, Brighina L, Begni B, Galbussera A, Difrancesco JC, et al. Alpha-synuclein nitration and autophagy response are induced in peripheral blood cells from patients with Parkinson disease. Neurosci Lett. 2010; 477: 6-10.
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002; 22: 782-790.
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008; 28: 7687-7698.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998; 392: 605-608.
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson's disease. Nature. 1998; 395: 451-452.
- McNaught KS, Olanow CW. Proteolytic stress: a unifying concept for the etiopathogenesis of Parkinson's disease. Ann Neurol. 2003; 53: S73-84.
- McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004; 56: 149-162.
- Zhang NY, Tang Z, Liu CW. alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem. 2008; 283: 20288-20298.
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997; 12: 25-31.
- Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002; 269: 1996-2002.
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005; 40: 622-633.
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009; 4: e5515.
- Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013; 23: 64-71.
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010; 8: e1000298.
- Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One. 2010; 5: e9367.
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004; 305: 1292-1295.
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010; 285: 13621-13629.
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010; 67: 1464-1472.
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008; 118: 777-788.
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009; 323: 124-127.
- Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008; 283: 23731-23738.
- Kabuta T, Wada K. Insights into links between familial and sporadic Parkinson's disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy. 2008; 4: 827-829.
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013; 16: 394-406.
- Pan PY, Yue Z. Genetic causes of Parkinson's disease and their links to autophagy regulation. Parkinsonism Relat Disord. 2014; 20 Suppl 1: S154-157.
- Sala G, Stefanoni G, Arosio A, Riva C, Melchionda L, Saracchi E, et al. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson's disease. Brain Res. 2014; 1546: 46-52.
- Rodriguez-Navarro JA, Cuervo AM. Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy. 2012; 8: 1152-1154.
- De Anda-Hernández MA, Lira-De León KI, Mena R, Campos-Peña V, Meraz-Ríos MA.Tau and Amyloid-ß Conformational Change to ß -Sheet Structures as Effectors in the Development of Alzheimer's Disease. Dr. Carlos M. Contreras, editor. In: Neuroscience - Dealing With Frontiers. InTech. 2012; 363-388.