
Research Article
Austin Alzheimers J Parkinsons Dis (aapd). 2023; 6(1): 1034.
Investigating the Involvement of Cytokines and Neurotrophic Factors in the Advanced Stages of Huntington’s Disease: A BACHD Study
Priscila Aparecida Costa Valadão¹*; Bruna da Silva Oliveira¹; Caroline Amaral Machado¹; Heliana de Barros Fernandes¹; Thatiane Cristina Machado¹; Kívia Santos Soares¹; Antonio Lucio Teixeira2,3; Cristina Guatimosim¹; Aline Silva de Miranda¹*
¹Departamento de Morfologia, Universidade Federal de Minas Gerais, Belo Horizonte –MG, Brazil
²Neuropsychiatry Program, Department of Psychiatry and Behavioral Sciences, McGovern Medical School, University of Texas Health Science Center at Houston, USA
³Faculdade Santa Casa BH, Belo Horizonte, MG, Brazil
*Corresponding author: Aline Silva de MirandaDepartamento de Morfologia, ICB Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Belo Horizonte, MG 31270-901 Brazil. Email: drapriscilavaladao@gmail.com; mirandaas@icb.ufmg.br; mirandas.aline@gmail.com
Received: February 04, 2023 Accepted: March 27, 2023 Published: April 03, 2023
Abstract
Neuroinflammation seems to be involved in the pathophysiology of Huntington’s Disease (HD), but its specific role on different stages of the disease, especially in later stages, remains to be understood. Here in, we investigated the concentrations of cytokines, chemokines and neurotrophic factors in striatum and frontal cortex of 24-month-old BACHD mice, a murine model of that displays several behavioral and pathological features of human HD. Our results revealed increased concentrations of the chemokine MCP-1 and the neurotrophin NGF in the striatum of BACHD mice alongside a reduction in the levels of the cytokine IL-6 and of the neurotrophin BDNF. In the frontal cortex, we found decreased levels of BDNF and MCP-1. We provide the first evidence that cytokines and neurotrophic factors may contribute to the pathophysiology of advanced HD.
Keywords: Huntington’s disease; Neuroinflammation; Neurotrophic factors; Cytokines; Chemokines
Introduction
Huntington's Disease (HD) is an autosomal-dominant inherited neurodegenerative disease caused by abnormal expansion of CAG trinucleotide repeats in the Huntingtin gene (HTT) located on chromosome 4 (The Huntington's Disease Collaborative Research Group, 1993; [2]. This mutation promotes huntingtin misfolding and formation of insoluble toxic aggregates in the nucleus and cytoplasm of neurons. In the Central Nervous System (CNS), the expression of mutant huntingtin proteins (mHTT) has also been described in microglia and astrocytes (Van der Burg et al., 2009; Jansen et al., 2017). Accumulations of mHTT with subsequent atrophy of brain areas such as striatum, cerebral cortex, hippocampus and brain stem, contribute to the constellation of motor, cognitive and psychiatric symptoms observed in HD [26,27,33]. As the progression of HD symptoms occurs in parallel with the neuropathological changes, HD seems to be a unique model to investigate neurobiological mechanisms involved in neurodegeneration [7,8,17].
Apart from the well-known role of mHTT proteins in HD development, the cellular and molecular mechanisms underlying disease progression remain to be fully understood. Accumulating evidence has supported a pivotal role of neuroinflammation in HD pathophysiology (Björkqvist et al., 2008) [7,9,35]. For instance, post-mortem studies demonstrated higher levels of inflammatory mediators such as IL-6, IL-8, tumor necrosis factor-α (TNF-α), monocyte chemoattractive protein-1 (MCP-1)/CCL2 and the anti-inflammatory cytokine IL-10 in the striatum, cortex and cerebellum of HD patients compared to controls (Björkqvist et al., 2008) [32]. Similar findings have been also reported in pre-clinical studies with rodent models of HD [14,15,23,29].
Neurotrophic factors, such as BDNF, NGF and GDNF, are a group of growth factors that play important roles in neuronal survival, differentiation and plasticity [20]. In parallel with HD-associated neurodegeneration, BDNF deficits have been reported in cell lines expressing mHTT and in brains of HD murine models and patients (FERRER et al., 2000; SEO et al., 2004;) [11,37]. Up-regulation of BDNF and GDNF in the striatum leads to neuroprotective effects in the YAC72 mice, a transgenic model of HD with over expression of mutant full-length huntingtin [37], as well as in a toxic model of HD induced by quinolinic acid (Martınez-Serrano & Bjorklund, 1996; Perez-Navarro et al., 1996; Alberch, 1999) [37].
Due to ethical concerns associated with human brain studies, animal models, especially genetic modified murine models, have been valuable tools to investigate HD-related pathophysiological mechanisms [24,35]. The BACHD transgenic mouse model carries the full-length human HTT gene in bacterial artificial chromosome and displays behavioral and neuropathological features of the human disease (Gray et al., 2008). Compared with other transgenic models of HD like the R6/2 mice, the BACHD mice live more than 24 months and exhibit a slow progression of the disease, which allows the study of pathophysiological mechanisms implicated in HD advanced stages (Gray et al., 2008; Yang, 1997) [19].
We hypothesize that an imbalance between inflammatory cytokines and neurotrophic factors may underlie HD progression. Here, using the BACHD murine model, we investigated the expression of cytokines and neurotrophic factors in 24-month-old mice. To the best of our knowledge, this is the first study addressing the involvement of these mediators in advanced HD. Our findings may shed lights in potential mechanisms involved in HD progression opening new venues for the identification of novel therapeutic targets.
Materials and Methods
BACHD mice
All experiments were carried out in accordance with the rules applicable by the local authorities (Ethics Committee on Animal Experiments of the Universidade Federal de Minas Gerais-CEUA/UFMG; approved protocol #036/2013). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Male FVB/NJ (wild-type or WT) and FVB/N-Tg (HTT*97Q)IXwy/J (BACHD) transgenic mice were purchased from Jackson Laboratory (Barl Harbor, ME, USA) (JAX stock #008197) and used to establish a colony. Food and water were offered ad libitum and the mice were kept in a temperature-controlled place (23°C) in a 12 to 12 hour light-dark cycle. All animals used in this study were genotyped ten days after birth using multiplex Polymerase Chain Reaction (PCR) (HTT-Forward: CCGCTCAGGTTCTGCTTTTA/HTT-Reverse: GTCGGTGCAGCGGCTCCTC; Actin-Forward: TGGAATCGTGTGGCATCCATCA/Actin-Reverse: AATGCCTGGGTACATGGGGTA). The animals were identified by numbers according to their genotype (WT or BACHD) and subsequently separated in mini-isolation cages with a maximum of 4 animals per cage.
All experiments were conducted with 24-month-old WT and BACHD mice as this age corresponds to older age in humans. Accordingly, the 24-month-old BACHD animals have established neurodegeneration, allowing the investigation of cytokine and neurotrophic factors alterations in the main brain areas affected by HD (Flurkey, Currer, and Harrison, 2007; Gray et al., 2008). Our research group has conducted several studies with male BACHD mice [16,35], as HD-associated behavioral phenotypes do not seem to be influenced by sex in this model [19].
Assessment of Cytokines, Chemokines and Growth Factors in Brain Regions
Brain regions (striatum and prefrontal cortex) of WT and BACHD mice (n=5 per group) were carefully removed and homogenized in an extraction solution (100mg of tissue per milliliter), containing 0.4MNaCl, 0.05%Tween 20, 0.5%BSA, 0.1mM phenyl methyl sulphonyl fluoride, 0.1mM benzethonium chloride, 10mM EDTA, and 20KIU aprotinin, using Ultra-Turrax. The lysates were centrifuged at 13.000×g for 10 min at 4°C and supernatants were collected and stored −70°C until use. The brain concentrations of the cytokines IL-6, IL-12p70, TNF-α and of the chemokine MCP-1/CCL2 were measured using an inflammatory CBA kit (BD Biosciences, San Diego, CA) and acquired on a FACS CANTO II flow cytometer (Becton Dickinson, San Jose, CA). These cytokines have been implicated in human HD development and/or progression (Björkqvist et al., 2008; [10,32]. The CBA results were analyzed by employing the software FCAP Array version 3.0 (Soft Flow Inc. Pecs, Hungary). The concentration of the neurotrophic factors, Brain-Derived Neurotrophic Factor (BDNF), Nerve Growth Factor (NGF), Glial cell-Derived Neurotrophic Factor (GDNF) and of the chemokine Fractalkine/CX3CL1 was determined by ELISA (R&D Systems, Minneapolis, MN) in accordance to the manufacturer’s instructions. Results are expressed as picogram per 100mg of tissue. The detection limit of the CBA and ELISA assays was 0.2 and 5pg/mL, respectively.
Statistical Analysis
All results are presented as mean±SD. The Shapiro-Wilk test was used in all data to determine the normality. For variables normally distributed differences were compared by Student's t-test. In case of variables not normally distributed differences were analyzed by Mann–Whitney U test. The significance was set at p<0.05. Statistical analyses were performed using Prism 6 software (GraphPad, La Jolla, CA, USA).
Results
At 24-month-old, has already been demonstrated in work previously published by our research group that BACHD mice display significant motor deficits as revealed by a decrease in the total distance traveled in the open field (p=0.0003) and in the latency to fall in both wire-hang (p=0.0073) and rotarod (p=0.0043) compared with controls [16].
In the striatum, BACHD mice presented decreased concentration of IL-6 (BACHD: 5.618±0.4367 Vs. WT: 6.833±0.1653; **p=0.04; Figure 1E) and BDNF (BACHD: 148.6±9.075 Vs. WT: 218.6±18.66; **p=0.008; (Figure 1G) and increased concentration of MCP-1 (BACHD: 59.91±4.762 Vs. WT: 36.23±7.436; *p=0.03; (Figure 1D) and NGF (BACHD: 4900±530.8 Vs. WT: 2934±416.9; *p=0.02; (Figure 1H) compared with WT animals. No significant differences were found in the concentrations of IL-12p70(p=0.60), TNF-α(p=0.91), GDNF(p=0.93) and CX3CL1(p=0.95).
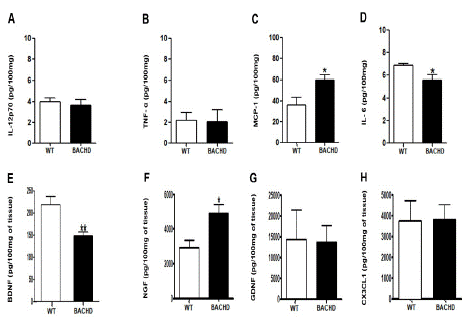
Figure 1: Increased MCP-1 and NGF and decreased IL-6 and BDNF concentration in the striatum of BACHD animals at 24-months-old. The figure 1 A-H shows the graphical quantification of cytokines, chemokines and neurotrophic factors concentration in the WT and BACHD mice striatum. The results are shown as mean±SD from n=5 animals per genotype. (MCP-1: *p=0.03; IL-6: **p=0.04; BDNF: **p=0.008; NGF: *p=0.02).
BACHD mice also showed decreased concentrations of MCP-1 (BACHD: 22.40±3.661 Vs. WT: 37.10±2.855; *p=0.03; (Figure 2D) and enhanced levels of BDNF in the prefrontal cortex compared with WT (BACHD: 135.5±15.55 Vs. WT: 92.73±9.904; *p=0.04; (Figure 2G). No significant changes were found in the concentration of IL-12p70(p=0.92), TNF-α(p=0.18), NGF(p=0.28), GDNF(p=0.33) and CX3CL1(p=0.39).
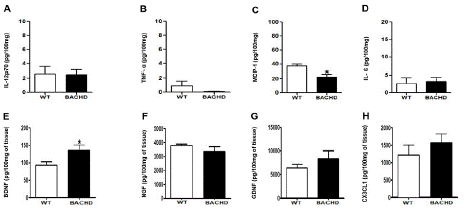
Figure 2: Increased BDNF and decreased MCP-1 concentration in the prefrontal cortex of BACHD animals at 24-months-old. The figure 1 A-H shows the graphical quantification of cytokines, chemokines and neurotrophic factors concentration in the WT and BACHD mice in the prefrontal cortex. The results are shown as mean±SD from n=5 animals per genotype. (MCP-1: *p=0.03; BDNF: *p=0.04).
Discussion
In the current study, we showed that BACHD mice at 24-months old that corresponds to the final stage of HD, displayed an imbalance in the levels of inflammatory mediators (IL-6 and MCP-1) and neurotrophic factors (BDNF and NGF) in key-brain areas, especially the striatum (Figure 3). The disruption in cytokines and neurotrophic factors signaling may contribute to HD-associated neurodegeneration. Increased levels of inflammatory cytokines have been implicated in HD pathophysiology [9]. However, the involvement of IL-6 remains to be further clarified as preclinical studies in rodent models have supported both protective and negative roles for increased IL-6 actions in HD (Bjorkqvist et al., 2008) [3,4,18]. We found decreased IL-6 levels in the striatum of BACHD mice. In a toxic model of HD induced by quinolinic acid, administration of IL-6 or the chimeric molecule IL6/IL6R, in which IL-6 and its soluble receptor are fused, in the striatum of Wistar rats improved striatal damage [3]. A more recent study also supports a protective effect of IL-6 in HD as the genetic deletion of IL6 in the R6/2 mice, a well-established HD transgenic model, exacerbated HD-related motor symptoms in comparison with R6/2 mice without the IL-6 deficiency [18]. Taken together these studies support our findings, as decreased striatal concentrations of IL-6 were associated with significant motor impairment in BACHD mice.
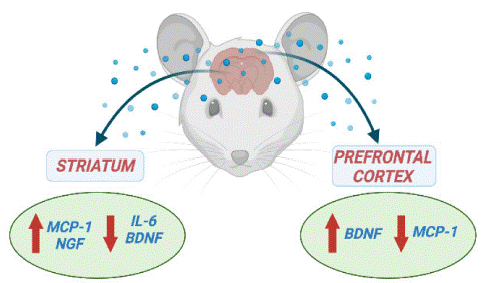
Figure 3: Cytokine and neurotrophic factors levels in the striatum and prefrontal cortex in the endpoint of Huntington’s Disease.
BACHD mice also displayed reductions of the neurotrophin BDNF in the striatum compared with controls. Significant decrease of BDNF concentration was also reported in the striatum of HD patients (Ferrer et al., 2000) as well as in the striatum of 20-week old Hdh Q111 mice, an Htt knock-in murine model of HD (Gines et al., 2003). Conversely, we found enhanced levels of BDNF in the prefrontal cortex of BACHD mice compared with controls. BDNF is a key component of the cortico-striatal synapse activity (Zuccato et al., 2005). It is mostly of cortical origin and anterogradely transported to the striatum (Altar et al., 1997; Baquet et al., 2004) [37]. It is quite tempting to hypothesize that the increase in BDNF in the prefrontal cortex of BACHD mice might be a compensatory effort to maintain BDNF supply to the striatum. The suppression of cortical BDNF via conditional mutagenesis in mice leading to significant impairment of striatal medium-spiny neurons along with clasping of hind limbs and inability for complex learning, symptoms present in HD, [6,30] (Baquet et al., 2004), support our hypothesis. Impairment in the delivery of BDNF from cortex to striatum have been also reported in primary cultures of cortical neurons [11].
Apart from the reduction of BDNF concentration in the striatum, we found enhanced levels of the neurotrophin NGF in this brain area. Higher mRNA expression and protein levels of NGF was also reported in the striatum after intrastriatal injection of quinolinic acid in rats. Increase of NGF levels protected striatal cholinergic interneurons from damage, suggesting an endogenous neuroprotective mechanism [1,5,21]. Transplantation of human-derived adipose stem cells secreting NGF in the striatum of YAC128 mice reduced striatal atrophy and improved motor performance in the rotarod task, corroborating this assumption [13]. Similar findings were also reported after transplantation of NGF-secreting stem cells in the striatum of rats before the injection of quinolinic acid (Martínez-Serrano & Bjorklund. In this scenario, the increase in NGF levels might be a compensatory phenomenon to prevent the striatal damage in advanced stage of HD.
Regarding chemokine expression, our study revealed increased levels of MCP-1 in the striatum while reduced levels were found in the prefrontal cortex. MCP-1 is one of the key chemokines that regulate the recruitment and activation of monocytes and microglia in a lesion site (Deshmane et al., 2009). MCP-1 signaling in the central nervous system has been implicated in neurodegenerative conditions such as Alzheimer’s disease and Parkinson’s disease (Chen et al., 2018) [28]. However, the available evidence on its involvement in HD is still limited. A post-mortem study revealed higher mRNA expression of MCP-1 in the striatum but not in the cortex of HD patients compared with controls [32]. Increased plasma levels of MCP-1 were associated with HD progression (Wild et al., 2009). Enhanced expression of MCP-1 was reported in mouse neuroblastoma cells expressing mHTT [12]. Further studies are necessary to explore the significance of our data regarding MCP-1 response in different brain regions in the context of HD.
We provided original evidence that cytokines, chemokine and neurotrophic factors may contribute, in a region-dependent manner, to the pathophysiology of advanced HD. Our findings may help to broaden our knowledge regarding the neurobiological processes underlying HD progression.
Author Statements
Conflicts of Interest
The authors declare no conflict of interest.
Author Contributions
PACV participated in the experimental design, carried out immunological assays, data analysis and drafted the manuscript. BSO and CAM participated in the experimental design, carried out immunological assays, data analysis and revised the manuscript. HBF, TGM and KS carried out immunological assays and revised the manuscript. ALT, CG and ASM participated in the design and coordination of the study, in the interpretation of experiments and editing the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This study was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). ALT, CG and ASM are supported by Brazilian National Council for Scientific and Technological Development (CNPq–bolsa de produtividade em pesquisa).
Institutional Review Board Statement
The study was conducted according to the committee that regulates the use of laboratory animals (Ethics Committee on Animal Experiments-CEUA/UFMG), which approved all experimental protocols under protocol #036/2013.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Alberch J Pérez-Navarro, Esther Canal J. Neurotrophic factors in Huntington’s disease. Prog Brain Res. 2004; 146: 195-229.
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, et al. Huntington disease. Nat Rev Dis Prim. 2015; 1: 15005.
- Bensadoun JC, de Almeida LP, Dreano M, Aebische rP, Deglon N. Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in the quinolinic acid rat model of Huntington’s syndrome. Eur J Neurosci. 2001; 14: 1753–61.
- Bouchard J, Truong J, Bouchard K, Dunkelberge rD, Desrayaud S, et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J Neurosci. 2012; 32: 18259–68.
- Canals JM, Marco S, Checa N, Michels A, Pe ́ rez- Navarro E. et al. Differential regulation of the expression of NGF, BDNF and NT-3 after excitotoxicity in a rat model of Huntington’s disease. Neurobiol Dis. 1998; 5: 357–364.
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999; 19: 3248–57.
- Colpo GD, Rocha NP, Stimming EF, Teixeira AL. Immunomodulatory Strategies for Huntington’s Disease Treatment. CNS Neurol Disord Drug Targets. 2017b; 16: 936–944.
- Colpo GD, Stimming EF, Rocha NP, Teixeira AL. Promises and pitfalls of immune-based strategies for Huntington’s disease. Neural Regen Res. 2017a; 12: 1422–1425.
- de Oliveira Furlam T, Roque IG, da Silva EWM, Vianna PP, Valadão PAC, et al. Inflammasome activation and assembly in Huntington’s disease. Molecular Immunology. 2022; 151: 134-142.
- Dobson L, Träger U, Farmer R, Hayardeny L, Loupe P, et al. Laquinimod dampens hyperactive cytokine production in Huntington’s disease patient myeloid cells. J Neurochem. 2016; 137: 782-94.
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2014; 118: 127-138.
- Godavarthi SK, Narender D, Mishra A, Goswami A, Rao SN, et al. Induction of chemokines, MCP‐1, and KC in the mutant huntingtin expressing neuronal cells because of proteasomal dysfunction. Journal of neurochemistry. 2009; 108: 787-795.
- Im W, Lee ST, Park JE, Shim J, Lim J, et al. Transplantation of patient-derived adipose stem cells in YAC128 Huntington’s disease transgenic mice. PLoS currents. 2010; 2: RRN1183.
- Kalonia H, Kumar P, Kumar A. Attenuation of proinflammatory cytokines and apoptotic process by verapamil and diltiazem against quinolinic acid induced Huntington like alterations in rats. Brain research. 2011; 1372: 115-126.
- Kalonia H, Mishra J, Kumar A. Targeting neuro-inflammatory cytokines and oxidative stress by minocycline attenuates quinolinic-acid-induced Huntington’s disease-like symptoms in rats. Neurotoxicity research. 2012; 22: 310-320.
- Kangussu LM, Rocha NP, Valadão PA, Machado TC, Soares KB. Et al. Renin-Angiotensin System in Huntington′ s Disease: Evidence from Animal Models and Human Patients. International Journal of Molecular Sciences. 2022; 23: 7686.
- Kim A, Lalonde K, Truesdell A, Gomes Welter P, Brocardo PS, et al. New Avenues for the Treatment of Huntington’s Disease. Int J Mol Sci. 2021; 22: 8363.
- Mary H Wertz, S Sebastian Pineda, Hyeseung Lee, Ruth Kulicke, Manolis Kellis, et al. Myriam Heiman. Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol Neurodegener. 2020; 15: 29.
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiology of disease. 2009; 35: 319-336.
- Padmakumar S, Taha MS, Kadakia E, Bleier BS, Amiji MM. Delivery of neurotrophic factors in the treatment of age-related chronic neurodegenerative diseases. Expert Opinion on Drug Delivery. 2020; 17: 323-340.
- Pérez-Navarro E, Alberch J, Arenas E, Calvo N, Marsal J. Nerve growth factor and basic fibroblast growth factor protect cholinergic neurons against quinolinic acid excitotoxicity in rat neo striatum. Eur J Neurosci. 1994; 6: 706–711.
- Plinta K, Plewka A, Pawlicki K, Zmarzły N, Wójcik-Pędziwiatr M, et al. The Utility of BDNF Detection in Assessing Severity of Huntington’s Disease. Journal of Clinical Medicine. 2021; 10: 5181.
- Ramachandran S, Thangarajan S. Thymoquinone loaded solid lipid nanoparticles counteracts 3-Nitropropionic acid induced motor impairments and neuroinflammation in rat model of Huntington’s disease. Metab Brain Dis. 2018; 33: 1459-1470.
- Ribeiro FM, Camargos ERS, de Souza LC, Teixeira AL. Animal models of neurodegenerative diseases. Rev Bras Psiquiatr. 2013; 35: s82-91.
- Rocha NP, Charron O, Latham LB, Colpo GD, Zanotti-Fregonara P, et al. Microglia Activation in Basal Ganglia Is a Late Event in Huntington Disease Pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2021; 8: e984.
- Rüb U, Hentschel M, Stratmann K, Brunt E, Heinsen H, et al. Huntington’s disease (HD): degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol. 2014; 24: 247–260.
- Rüb U, Hoche F, Brunt ER, Heinsen H, Seidel K, et al. Degeneration of the cerebellum in Huntington’s disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol. 2013; 23: 165–177.
- Santaella A, Kuiperij HB, van Rumund A, Esselink RA, van Gool AJ, et al. Inflammation biomarker discovery in Parkinson’s disease and atypical parkinsonisms. BMC neurology. 2020; 20: 26.
- Saroj P, Bansal Y, Singh R, Akhtar A, Sodhi RK, et al. Neuroprotective effects of roflumilast against quinolinic acid-induced rat model of Huntington’s disease through inhibition of NF-κB mediated neuroinflammatory markers and activation of cAMP/CREB/BDNF signaling pathway. Inflammo pharmacology. 2021; 29: 499-511.
- Schmidtke K, Manner H, Kaufmann R, Schmolck H. Cognitive pro- cedural learning in patients with fronto-striatal lesions. Learn Mem. 2002; 9: 419–29.
- Serrano MA, Bjorklund A. Protection of the neostriatum against excitotoxic damage by neurotrophin- producing, genetically modified neural stem cells. J Neurosci. 1996; 16: 4604-4616.
- Silvestroni A, Faull RL, Strand AD, Möller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009; 20: 1098-1103.
- Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron. 2019; 101: 801–819.
- Tripathy D, Yin X, Sanchez A, Luo J, Martinez J, et al. Cerebrovascular expression of proteins related to inflammation, oxidative stress and neurotoxicity is altered with aging. Journal of Neuroinflammation. 2010; 7: 63.
- Valadão PAC, Santos K, Ferreira E Vieira TH, Macedo E Cordeiro T, Teixeira AL, et al. 2020. Inflammation in Huntington’s disease: A few new twists on an old tale. J Neuroimmunol. 2020; 348: 577380.
- Wertz MH, Pineda SS, Lee H, Kulicke R, Kellis M, et al. Heiman M. Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol Neurodegener. 2020; 15: 29.
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001; 293: 493–498.