
Special Article: Movement Disorders
Austin Alzheimers J Parkinsons Dis. 2023; 6(1): 1038.
69-Year-Old Female with Progressive Supranuclear Palsy, Frontotemporal Dementia, Focal Hyperkinesia and one variant in the GCH1 gene and in the CSF1R gene
Panteleimon Oikonomou¹*; Sabine Hellwig²; Thomas Wehrum²; Horst Urbach³; Wolfgang H Jost¹
¹Center for Movement Disorders, Parkinson-Klinik Ortenau, Wolfach, Germany
²Department of Psychiatry and Psychotherapy Medical Center - University of Freiburg, Germany
³Department of Neuroradiology, University Medical Center Freiburg, Faculty of Medicine, University of Freiburg, Germany
*Corresponding author: Panteleimon Oikonomou Center for Movement Disorders, Parkinson-Klinik Ortenau, Wolfach, Germany. Email: p.oikonomou@parkinson-klinik.de
Received: March 20, 2023 Accepted: April 28, 2023 Published: May 05, 2023
Abstract
How insignificant is the detection of gene variants in a novel clinical presentation of an atypical Parkinson’s syndrome with positive family history? We present the case of a 69-year-old woman with Progressive Vertical Supranuclear Palsy (PSP), focal hyperkinesia, non-L-dopa-responsive asymmetrical Parkinsonism, postural instability as well as apraxia, cognitive, speech and frontal disorder, with a positive family history, MRI evidence of mesial temporal atrophy and tau-PET evidence of tau pathology as well as identification of one variant in the GCH1 gene and one variant in the CSF1R gene. The fact that the clinical presentation and radiological finding of our patients are not compatible with a known and well described genetic disorder caused by pathogenic mutations of these genes, supports the notion that the detected gene variants have an uncertain significance in the pathogenesis of our patient’s disease.
Nevertheless, the novel description of the phenotype of our case of PSP with frontal lobe features, focal hyperkinesia and a positive family history, points to an in-vivo tauopathy, in which a genetic burden may play a role. The follow-up of the patient und her family may offer further insights into the pathophysiology of complex neurodegenerative diseases in the spectrum of tauopathies, offering possible new therapeutic targets.
Abbrevations: PSP: Progressive Supranuclear Palsy; FTD: Frontotemporal Dementia; CBS: Corticobasal Syndrome; GCH1: Guanosine 50-Triphosphate Cyclohydrolase I; GTPCH1: GTP Cyclohydrolase 1; LRD: L-Dopa-Responsive Dystonia. PD: Parkinson Disease; HDLS: Hereditary Diffuse Leukoencephalopathy with Spheroids; I-123-FP-CIT; SPECT: N-(3-Fluoropropyl)-2β-Carbomethoxy-3β-(4-[123I]iodophenyl) Nortropane Single-Photon Emission Computed Tomography. FDG-PET: Fluorodeoxyglucose-Positronemission Tomography; CSF: Cerebrospinal Fluid; NGS (Next-Generation Sequencing).
Introduction
Progressive Supranuclear Palsy (PSP) is a rare neurodegenerative disorder, with prominent 4R-tau neuropathology, a variety of clinical presentations due to an effect on four clinical domains (ocular motor dysfunction, postural instability, akinesia, cognitive impairment) [1], and a spectrum of overlapping phenotypic subtypes from the Richardson-syndrome to variants such as PSP with parkinsonism, speech disorder, Frontotemporal Dementia (FTD) and Corticobasal Syndrome (CBS) [2]. Though PSP is generally regarded as a sporadic disorder, there is increasing evidence suggesting that a series of common and rare genetic variants have an impact on sporadic and familial forms of PSP [3]. A PSP-like phenotype with focal hyperkinesia, not associated with PSP-CBS and drug-induced hyperkinetic disorder has not been described before to our knowledge.
Guanosine 50-triphosphate cyclohydrolase I (GCH1) is located on chromosome 14 (14q22) and encodes the enzyme GTP cyclohydrolase 1 (GTPCH1) [4]. Loss-of-function mutations in GCH1 gene have been shown to cause autosomal recessive GCH-deficient hyperphenylalaninemia and autosomal dominant L-Dopa-Responsive Dystonia (LRD) [5]. Although LRD is a rare neurometabolic disease, characterized with dystonia that begins in childhood and responds dramatically and permanently to Low-Dose Llevodopa (L-dopa), an association with Parkinsonism in old age has been reported [4]. Furthermore, patients with sporadic Parkinson Disease (PD) have an increased frequency of pathogenic GCH1 mutations, which have previously been reported to cause LRD [5]. Thus, rare GCH1 coding variants are considered as a risk factor for PD [6].
Colony stimulating factor 1 receptor (CSF1R) gene is located on chromosome 5 (5q32) and encodes a cell-surface receptor primarily for the cytokine CSF-1, which controls the production, differentiation, and function of macrophages [7]. Mutations affecting the tyrosine kinase domain of CSF1R cause Hereditary Diffuse Leukoencephalopathy with spheroids (HDLS) [8,9]. Possible roles of CSF1R in neurodegenerative diseases including PD have been proposed [7].
Case Description
A 69-year-old female patient presented in our clinic with a history of slowly progressive symptoms since approximately one year including initial visual disturbances i.e. diplopia, and later an inability to make vertical eye movements combined with dizziness and headaches, involuntary movements of the left arm, an unsteady shuffling gait with falls, and amnestic aphasia. According to her husband, the patient's personality und behaviour showed alterations including irritability and emotional instability. In addition, cognitive changes such as concentration and memory impairment, as well as dysfunction of rational thinking and executive functions were also present. Everyday life activities were hence restricted with a heavy dependency on her husband.
A positive family-history was reported. The patient´s brother had "brain damage", unknown diagnoses and died at the age of 60. No neurological issues were identified in the children and the parents of the patient.
A detailed neurological examination revealed vertical supranuclear palsy viewing up, involuntary exploratory stereotypic choreiform movements of the distal upper left extremity with dystonic component, hypo- and bradykinesia mainly left-sided, pronounced postural instability, applause sign, non-fluent aphasia, idiomotoric apraxia, as well as executive (abnormal Luria-test) and behavioral dysfunction with affective lability.
Diagnostic Assessment
The diagnostic assesments were performed in the University Medical Center Freiburg. The Magnetic Resonance Imaging (MRI) of the head showed mesiotemporal atrophy including entorhinal atrophy and minor atrophy seen in the superior vermis more pronounced on the right side (Figure 1). N-(3-Fluoropropyl)-2β-carbomethoxy-3β-(4-[123I]iodophenyl) nortropane single-photon emission computed tomography (I-123-FP-CIT SPECT) revealed a slightly reduced availability of dopamine transporters in the right striatum (Figure 2).
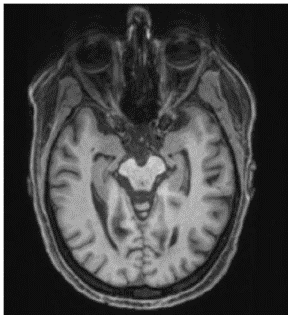
Figure 1: MRI of the head showed mesiotemporal atrophy more pronounced on the right side.
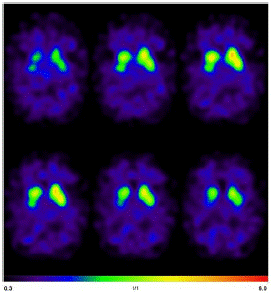
Figure 2: I-123-FP-CIT SPECT revealed a slightly reduced availability of dopamine transporters in the right striatum.
Fluorodeoxyglucose-Positron Emission Tomography (FDG-PET) provided evidence of moderate to slight hypometabolism on the right mesial and high dorsolateral frontal, tapering anteriorly along the premotor cortex, and less in the bilateral caudate nucleus (Figure 3). Additionally, tau-PET was performed and showed a slight to moderate tau pathology highly dorsolateral anteriorly, as well as bilateral mesencephalic, thalamic to pallidostriatal emphasized on the right side (Figure 4).
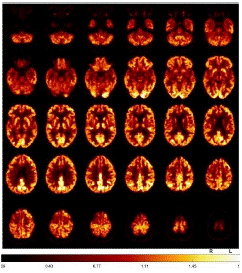
Figure 3: FDG-PET demonstrated a moderate to slight hypometabolism on the right mesial and high dorsolateral rontal, tapering anteriorly along the premotor cortex, and less in the bilateral caudate nucleus.
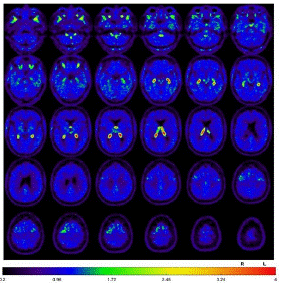
Figure 4: Tau-PET, slight to moderate tau pathology highly dorsolateral anteriorly, as well as bilateral mesencephalic, thalamic to pallidostriatal emphasized on the right side.
The relevant routine laboratory examinations were normal. Examination of the Cerebrospinal Fluid (CSF) revealed normal findings, with no evidence of pathological antineuronal antibodies. Furthermore, Whipple PCR, diagnostic tests for Wilson’s disease as well as genetic analysis for Huntington’s chorea were negative. Next-Generation Sequencing (NGS) panels for dystonia and PD revealed variants of unclear significance for the genes GCH1 (NM_000161.3, chr14:55310817, c.671A>G, p.K224R (het.)) and CSF1R (NM_005211.4, chr5:149449462, c.1484C>T, p.S295F (het.)).
Therapeutic Intervention
The symptoms were not L-Dopa-responsive, although treatment with amantadine 100mg/d showed slight improvement of hypo- and bradykinesia, of apraxia as well as the hyperkinesia. Supplementation with coenzyme Q103X 5mg/kg as a possible neuroprotective factor was initiated. Since Sertraline was not tolerated, initially 7.5mg/d of mirtazapine was prescribed against sleep disorders and affective lability.
At the same time we performed intensive physiotherapy focusing on balance and walking training in everyday situations, developing strategies for better body position changes, speech training focusing on differentiation of self-perception to increase the depth of breathing and articulation exercises as well as occupational therapies to improve fine motor skills, coordination, the mobility of the upper extremities and instructions for self-exercises and relaxation exercises.
Discussion
We present a case of a 69-year-old woman with vertical supranuclear palsy, focal hyperkinesia, asymmetrical Parkinson’s syndrome, postural instability as well as apraxia, cognitive, speech and frontal disorder, with a positive family history, as well as identification of 2 gene variants related to movement disorders and neurodegeneration.
Molecular imaging provided evidence of tau pathology and glucose hypometabolism located mainly frontal, mesencephalic and in basal ganglia pronounced on the right side corresponding with FTD-PSP, as well as reduced availability of dopamine transporters in the right striatum, pointing to a predominant left-sided Parkinson syndrome. The atrophy pattern in MRI of the brain corresponded to a PSP- und Alzheimer's-disease-like pathology. Laboratory examination of blood and CSF did not indicate a secondary aetiology. Both the clinical presentation and the diagnostic findings concur with FTD-PSP with apraxia and non-levodopa induced hyperkinesia. The criteria of CBS requiring one cortical symptom and one asymmetrical extrapyramidal symptom are also fulfilled, however a parietal atrophy was missing. Meanwhile, it is well recognized that the phenotypic manifestation of a primary tauopathy such as PSP, FTD or CBS commonly overlaps [10]. To our knowledge, PSP with abnormal involuntary movements in the context of PSP-CBS is rare [11], but so far has not been described with respect to non-levodopa induced choreiform hyperkinesia.
In our patient, a mutation of the GCH1 gene was detected. The wildtype GCH1gene comprises six exons, and more than 200 different mutations have so far been identified [4]. GTP-cyclohydrolase 1, encoded by GCH1, is a crucial enzyme for dopamine biosynthesis in nigrostriatal cells [12]. In families with GCH1 mutations, co-occurrence of LRD and Parkinsonism has been reported [4]. The detected heterozygous pK224R muta In our patient, a mutation of the GCH1 gene was detected. The wildtype GCH1gene comprises six exons, and more than 200 different mutations have so far been identified [4]. GTP-cyclohydrolase 1, encoded by GCH1, is a crucial enzyme for dopamine biosynthesis in nigrostriatal cells [12]. In families with GCH1 mutations, co-occurrence of LRD and Parkinsonism has been reported [4]. The detected heterozygous pK224R mutation was hypothesized as causing LRD. Nevertheless, in a cohort of 1,113 patients with PD, in which the entire GCH1-coding regions was sequenced, the p.K224R mutation was found in 3 patients with late onset (58, 64 years) PD without any reported dystonia or hyperkinesia and with reported levodopa-response in one of them [5]. At 68 years of age, our patient developed an asymmetrical Parkinson-Syndrome unresponsive to L-Dopa, focal hyperkinesia but no dystonia, correlated with tau-pathology and an asymmetrical nigrostriatal denervation of striatum. Interestingly, the p.K224R mutation was also detected in a patient with late onset (age at onset 82 years) asymmetrical tremor, levodopa response und a tauopathy histopathology consistent with the diagnosis of PSP, but concomitant with Lewy body pathology [12].
Moreover, in our patient a heterozygous mutation of CSF1R was detected, which is associated with the rare disease HDLS. Typically, HDLS patients present with FTD-like phenotype in their 40s–50’s, accompanied by motor symptoms, including pyramidal and extrapyramidal signs [13], which share common features with our patient, although the main radiological finding of extended white matter lesions, was not present in our case. More than 50 pathogenic variants have been reported in patients with HDLS, all located in the intracellular tyrosine-kinase domain of the CSF1R gene encoded by exons 12-22 [9,14]. The detected mutation (CSF1R c.1484C>T, p.S295F) in our patient is not listed as a known mutation or polymorphism in standard databases of clinical genomes (https://www.ncbi.nlm.nih.gov/clinvar), although other mutations in the loci NM_005211.4 are reported with uncertain or benign clinical significance regarding HDLS, which is expected, being outside the exons 12-22. Mutations of CSF1R gene have also been identified in a few patients with a behavioral variant of FTD [7] and in a patient clinically diagnosed with CBS, [8] indicating a role of CSF1R in tauopathies. To our knowledge, a connection between PSP and CSF1R-mutations has not been described before.
The fact that the clinical presentation of our patients is not compatible either with LRD or with HDLS, support the notion that the detected gene variants have an uncertain significance in the pathogenesis of our patients’ disease. Nevertheless, our case of PSP-FTD with focal hyperkinesia and a positive family history, points to an in-vivo tauopathy, in which a genetic burden of a mutation combination, or concurrent with possible pathogenic mutation may play a role. The follow-up of the patient and her family may offer further insights into the pathophysiology of complex neurodegenerative diseases in the spectrum of tauopathies, offering possible new therapeutic targets.
Patient Perspective
Upon discharge, the patient and her husband reported an improvement through the new medication and at the same time through the occupational therapy applications. The patient was able to use her left hand more purposefully in everyday life. There are still limitations in everyday life especially due to the visual und cognitive impairment. During a home visitation, she herself expressed the hope that more insights could be gained on the underlying mechanisms of her disorder and feedback could serve treatment strategies by increasing the awareness of the existence of this phenotype due the publication of this case report. In any case, a neurological follow-up with adjustment of the medication, if necessary as well as continuation of regularly activating therapy are planned.
Author Statements
Availability of Supporting Data
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study
Competing Interests
The authors declare that they have no competing interests.
Funding
The authors declare that they have received no funding regarding this work.
Authors' Contribution
PO: conception and drafting the manuscript. SH, TW and WJ: critical revision the manuscript. HU is the head of the department where the MRI was performed.
Acknowledgments
We thank Prof. Meyer, head of the department of nuclear medicine in University Medical Center Freiburg for his kind approval to use the photographic material developed in his department.
References
- Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017; 32: 853-864.
- Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017; 16: 552-563.
- Wen Y, Zhou Y, Jiao B, Shen L. Genetics of Progressive Supranuclear Palsy: A Review. J Parkinsons Dis. 2021; 11: 93-105.
- Lewthwaite AJ, Lambert TD, Rolfe EB, Olgiati S, Quadri M, et al. Novel GCH1 variant in Dopa-responsive dystonia and Parkinson’s disease. Parkinsonism Relat Disord. 2015; 21: 394-7.
- Rudakou U, Ouled Amar Bencheikh B, Ruskey JA, Krohn L, Laurent SB, et al. Common and rare GCH1 variants are associated with Parkinson’s disease. Neurobiol Aging. 2019; 73: 231.e1-231.e6.
- Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, et al. Parkinson’s disease in GTP cyclohydrolase 1 mutation carriers. Brain. 2014; 137: 2480-92.
- Hu B, Duan S, Wang Z, Li X, Zhou Y, et al. Insights Into the Role of CSF1R in the Central Nervous System and Neurological Disorders. Front Aging Neurosci. 2021; 13: 789834.
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011; 44: 200-5.
- Chen J, Luo S, Li N, Li H, Han J, et al. A Novel Missense Mutation of the CSF1R Gene Causes Incurable CSF1R-Related Leukoencephalopathy: Case Report and Review of Literature. Int J Gen Med. 2020; 13: 1613-1620.
- Ganguly J, Jog M. Tauopathy and Movement Disorders-Unveiling the Chameleons and Mimics. Front Neurol. 2020; 11: 599384.
- Zhang W, Mao W, Xu E, Chhetri JK, Chan P. Progressive supranuclear palsy presenting with hyperkinetic movement disorder and hemiplegic dystonia: a case report. Int J Neurosci. 2020; 130: 1278-1281.
- Guella I, Sherman HE, Appel-Cresswell S, Rajput A, Rajput AH, et al. Parkinsonism in GTP cyclohydrolase 1 mutation carriers. Brain. 2015; 138: e349.
- Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R-related leukoencephalopathy: A major player in primary microgliopathies. Neurology. 2018; 91: 1092-1104.
- Stabile C, Taglia I, Battisti C, Bianchi S, Federico A. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): update on molecular genetics. Neurol Sci. 2016; 37: 1565-9.