
Research Article
Austin J Anal Pharm Chem. 2014;1(2): 1010.
A Chiral HPLC-MS/MS Method for Simultaneous Quantification of Warfarin Enantiomers and its Major Hydroxylation Metabolites of CYP2C9 and CYP3A4 in Human Plasma
Ju W1, Peng K1, Yang S1, Sun H2, Sampson M2 and Wang MZ1*
1Department of Pharmaceutical Chemistry, the University of Kansas, USA
2Department of Pharmacy, University of North Carolina at Chapel Hill, USA
*Corresponding author: :Wang MZ, Department of Pharmaceutical Chemistry, The University of Kansas, 2095 Constant Ave, Lawrence, KS 66047, USA.
Received: August 07, 2014; Accepted: August 20, 2014; Published: August 22, 2014
Abstract
Warfarin is an oral anticoagulant that requires frequent therapeutic drug monitoring due to a narrow therapeutic window, considerable inters individual variability in drug response, and susceptibility to drug-drug and drug-diet interactions. Enantiomeric separation and quantification of warfarin enantiomers and clinically important major hydroxylation metabolites are essential for drug interaction studies and phenotypic characterization of CYP2C9 and CYP3A4, the major Cytochrome P450 (CYP) enzymes involved in warfarin metabolism. Here, we describe the development and validation of a chiral high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/ MS)-based quantification of R-warfarin, S-warfarin, S-7-hydroxywarfarin (the major CYP2C9metabolite) and (9R; 10S)-10-hydroxywarfarin (the CYP3A4 metabolite) in human plasma. Simple protein precipitation-based extraction showed good recovery of analyses (82.9 - 96.9%). The developed method exhibited satisfactory intra-day and inter-day accuracy and precision. The lower limits of detection were 0.25nM (or ~0.08 ng/ml) for the war far in enantiomers and 0.1nM (or ~0.04 ng/mL) for S-7-hydroxywarfarinand (9R; 10S)-10- hydroxywarfarin using only 50μL plasma during extraction. The validated method was successfully applied to analyze plasma samples obtained from a healthy human subject who enrolled in a clinical drug interaction study involving warfarin.
Keywords: Warfarin; Chiral separation; HPLC-MS/MS; Hydroxyl war far in; Protein precipitation extraction
Abbreviations
AUC: Area Under The Plasma Concentration-Time Curve; COD: Coefficient of Determination; CYP: Cytochrome P450; CV: Coefficient of Variation; DMSO: Dimethylsulfoxide; HPLC-MS/ MS: High Performance Liquid Chromatography-tandem mass spectrometry; INR: International Normalized Ratio; IS: Internal Standard; LLE: Liquid–Liquid Extraction; LLOQ: Lower Limit Of Quantification; LOD: Limit Of Detection; MRM: Multiple Reaction Monitoring; PPE: Protein Precipitation Extraction; PT: Prothrombin Time; QC: Quality Control; SD: Standard Deviation; SPE: Solid- Phase Extraction; S/N: Signal-to-Noise ratio; ULOQ: Upper Limit Of Quantification
Introduction
Warfarin is the most commonly prescribed oral anticoagulant in North America and is indicated for the prophylaxis and treatment of a number of serious thromboembolic disorders and complications [1]. It elicits an anticoagulant effect by interfering with the synthesis of vitamin K-dependent clotting factors in the liver via inhibition of the vitamin K epoxies reeducates complex subunit 1 (VKORC1) [2]. Warfarin has a narrow therapeutic index and displays considerable inter individual variability in dose requirements. To achieve and maintain optimal anticoagulant activity: warfarin efficacy is frequently monitored by the pro thrombin time (PT) or the Internationally Normalized Ratio (INR). Despite target INR monitoring and careful dose adjustment: the rate of warfarin-related bleeding events has not diminished [3,4]. Genetic factors including VKORC1: concomitant drugs/diet: and various disease states can impact on the efficacy of warfarin by changing its pharmacokinetics. Thus: sensitive and specific analytical methods to quantify warfarin and its clinically significant metabolites in various matrices: especially human plasma: may help to improve the understanding of warfarin pharmacology and detection of potential warfarin-drug/diet interactions.
Warfarin is a racemic mixture of R- and S-enantiomers. S-warfarin is more potent (3-5 times) pharmacologically than R-warfarin [5]: however S-warfarin clearance is greater than that of R-warfarin. Both enantiomers undergo extensive cytochrome P450 (CYP)-mediated metabolism in the liver. CYP2C9 catalyzes the 6- and 7-hydroxylation of S-warfarin: with S-7-hydroxywarfarin (S-7-OH-warfarin) being the predominant metabolite: and the 4’-hydroxylation of R-warfarin [6]. CYP3A4 regio- and stereo selectively catalyzes the 10-hydroxylation of R-warfarin to form (9R; 10S)-10-hydroxywarfarin [(9R; 10S)-10- OH-warfarin] [7]. Inhibition and induction of CYP2C9- and/or CYP3A4-mediated warfarin metabolism by co-administered drugs/diets the main culprit for many clinically important drug interactions [1]. Therefore: specific quantification of S-7-OH-warfarin (or S-6- OH-warfarin) and (9R; 10S)-10-OH-warfarin in plasma will enable phenotypic characterization of CYP2C9 and CYP3A4 catalytic activities during clinical drug-drug and drug-diet interaction studies.
Several analytical methods have been reported to achieve Enantiomeric separation of R-and S-warfarin and/or the stereoisomer’s of clinically important warfarin metabolites. Separation and quantification of R-and S-warfarin in human plasma has been demonstrated using various chiral High Performance Liquid Chromatography (HPLC) columns coupled with detection by UV [8- 12] or tandem mass spectrometry (MS/MS) [13]. However: chiral separation and quantification of hydroxylated warfarin metabolites has mainly focused on 7-OH-warfarinusing a chiral HPLC column coupled with UV [14]: fluorescence [15] or MS/MS detection [16]: or using capillary zone electrophoresis coupled with UV detection [17]. Although specific quantification of the CYP3A4-selective warfarin metabolite: (9R; 10S)-10-OH-warfarin: in human plasma using MS/ MS detection has been reported [18]: analyses were separated by combining phenyl based reverse phase column and chiral column with a complicated sample preparation and a long analytical time (17 min). In addition: previously reported analytical methods for the warfarin enantiomers in plasma involve either extraction using solid phase extraction (SPE) cartridges [8;16;18-20] or phase separation via liquid-liquid extraction (LLE) [9-11,13-15]. Extraction of war far in by Protein Precipitation (PPE) has only been used for microsomal metabolism studies: where protein concentrations are typically much lower (< 2 mg/mL) than in plasma [21,22]. In this study: we aimed to 1) develop a chiral HPLC-MS/MS-based method to quantify the warfarin enantiomers and two stereo isomeric metabolites: S-7- OH-warfarin and (9R; 10S)-10-OH-warfarin: in human plasma for CYP2C9 and CYP3A4 phenotyping with a short chromatographic procedure and 2) determine whether PPE could be used as an alternative extraction method for Enantiomeric analysis of warfarin and its major hydroxylated metabolites in human plasma.
Materials and Methods
Materials
R-warfarin: S-warfarin: racemic warfarin: warfarin metabolites (4’-: 6-: 7-: 8- and 10-OH-warfarin): and warfarin-d5 (deuterated phenyl ring; used as internal standard [IS]) were purchased from Toronto Research Chemicals Inc. (North York: ON: Canada). Ammonium acetate and dimethylsulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis: MO: USA). Optima-grade water: acetonitrile: methanol: and acetic acid was obtained from Fisher Scientific (Pittsburgh: PA: USA). Blank human plasma (collected in K2-EDTA tubes) was purchased from Innovative Research (Novi: MI: USA). Recombinant human CYP3A4 and CYP2C9 Supersomes™: microsomes prepared from baculovirus-infected insect cells expressing human CYP enzymes and NADPH-cytochrome P450 reductase: were purchased from BD Gentlest (Woburn: MA: USA).
Incubation with recombinant CYP3A4 and CYP2C9 enzymes
The metabolism of R- and S-warfarin by recombinant CYP3A4 and CYP2C9 was conducted according to a previously published protocol [22] with modifications. Briefly: incubation mixtures (50 μL) contained R- or S-warfarin (10 μM final concentration): recombinant CYP enzymes individually (50 pmol/mL): and 100 mM phosphate buffer (pH 7.4) containing 3.3mM MgCl2. Reactions were initiated by the addition of NADPH (1 mM final concentration) and allowed to proceed for 30 min at 37°C. Control incubations were conducted in the absence of NADPH. The reactions were stopped with 400 μL ice-cold methanol-water (7:1: v/v) and vortexes. Following centrifugation to pellet precipitated proteins: the supernatants were dried under nitrogen at 50°C for 45 misusing a 96-well micro plate evaporator (Model SPE Dry 96; Biotage: LLC: Charlotte: NC: USA) and the dried sample reconstituted with 250 μL methanol-water (15:85: v/v) prior to chiral HPLC-MS/MS analysis.
Preparation of calibration standards and quality controls
Individual stock solutions of R-war far in: S-war far in: racemic war far in: warfarin-d5: and each war far in metabolite (4’-: 6-: 7-: 8- and 10-OH-warfarin) were prepared in DMSO. Working standard mixtures were prepared by mixing and diluting the stock solutions of racemes warfarin:7-OH-warfarin: and 10-OH-warfarin.Calibration standards were prepared by spiking 1.0 μL working standard mixture into 49 μL blank human plasma to yield the following final concentrations: 0.25: 0.5: 1.25: 2.5: 5: 12.5: 25: 50: 125: 250: 500: 1250: 2500: and 5000 nM for R- and S-warfarin: and 0.05: 0.1: 0.25: 0.5: 1: 2.5: 5: 10: 25: 50: 100: 250: 500: and 1000 nM for S-7- OH-warfarin and (9R;10S)-10-OH-warfarin.Quality controls (QCs): prepared separately from individual stock solutions: had final plasma concentrations of 1.25: 2.5: 50: and 1250 nM for R- and S-warfarin: and 0.25: 0.5: 10: and 250 nM for S-7-OH-warfarin and (9R;10S)-10- OH-warfarin. Calibration standards were prepared in triplicate: while QCs in quadruplicate; all were processed as plasma samples before chiral HPLC-MS/MS analysis.
Sample preparation and HPLC-MS/MS analysis
Following thawing at room temperature: plasma samples were prepared using PPE rather than SPE or LLE. Specifically: plasma samples (50 μL) were mixed with 400 μl methanol-water (7:1: v/v) containing 30 nM warfarin-d5 as the IS. After overtaxing for 10 s: the samples were centrifuged at 2250 g for 15 min at 4°C to pellet precipitated proteins. The supernatants were evaporating dander nitrogen at 50°Cfor 45 min. The dried samples were reconstituted with 100 μL methanol-water (15:85: v/v) prior to chiral HPLC-MS/ MS analysis.
HPLC-MS/MS quantification of warfarin and its metabolites was performed on a Waters ACQUITY I-Class UPLC (operated at normal HPLC pressure) coupled to a Xevo TQ-S triple quadruple mass spectrometer equipped with an electro spray ionization source (Waters Corporation: Milford: MA: USA). Enantiomeric separation of warfarin and its metabolites was achieved with an Astec CHIROBIOTIC® V Chiral HPLC column (100 mm × 4.6 mm: 5 μm; Supelco: Inc: Bellefonte: PA) protected by an ACQUITY column in-line filter (0.2 μm; Waters Corporation).HPLC mobile phase (A) consisted of 100% (v/v) water with 5mM ammonium acetate (pH4.0: adjusted with acetic acid): while (B) consisted of100% (v/v) acetonitrile. The initial gradient began with 10% B and was held for 0.2 min. Mobile phase B increased linearly to 40% over 5 min and remained at 40% for 1 min. The system was re-equilibrated with 10% B for 2 min prior to the next injection. A flow rate of 0.8 mL/min was used throughout the study. The auto sampler was set at 6°C and the HPLC column heated at 50°C.The sample injection volume was 10 μL.
The mass spectrometer was operated under negative ion mode for multiple reaction monitoring (MRM) of warfarin and its metabolites. Typical instrument conditions were: capillary voltage: 0.50 kV; source offset: 50 V; desolation temperature: 500°C; desolation gas flow: 1000 L/h; cone gas flow: 150 L/h; nebulizer gas flow: 7.0 bar; and collision gas flow: 0.15 ml/min. Analyze-specific instrument parameters (i.e.: collision energy: capillary voltage: cone voltage) were optimized prior to analysis using the Intel Start™ auto-tune with infusion of analyze standards. The specific MRM transitions used for quantification were m/z 307.1 →161.0 for warfarin: m/z 312.2→ 255.1 for warfarin-d5: m/ z323.1→177.0 for 6-: 7- and 8-OH-warfarin: and m/z 323.1→ 250.3 for 10-OH-warfarin. Peak area ratios of analyze vs. IS were used to generate calibration curves and calculate analyze concentrations in plasma samples.
Method validation
Method selectivity was investigated by comparing HPLC-MS/MS chromatograms from blank human plasma and plasma spiked with standards at the Lower Limit of Quantification (LLOQ). To ensure MRM transitions for warfarin and 6-: 7-: 8-: and 10-OH-warfarin did not cross-detect 4’-OH-warfarin: chromatograms from plasma spiked with 4’-OH-warfarin (200 nM final concentration) also were compared.
The intra-day accuracy and precision of the method were determined by replicate analysis (n = 4) of the QC son the same day. The inter-day accuracy and precision were determined by replicate analysis of the QC son three separate days. Accuracy (%) was defined as the closeness of the average QC concentrations determined by the method to the true concentrations. Precision (Coefficient of Variation [CV]; %) was defined as the spread of the individual measures of multiple QC preparations.
The extraction recovery was determined by comparing peak area ratios of analyses vs. IS from QC samples that were spiked with analyze before extraction to ratios from blank plasma extracts spiked with known amounts of pure analyze post-extraction (represents 100% recovery).The extraction recovery was examined at three QC concentrations (2.5: 50: and 1250 nM for R- and S-warfarin; 0.5: 10: and 250 nM for S-7-OH-warfarin and (9R;10S)-10-OH-warfarin) with quadruplicate samples at each concentration.
Matrix effects during MS/MS analysis were evaluated by comparing peak areas of analyze from blank plasma extracts spiked with known amounts of pure analyze to peak areas from samples prepared in the reconstitution solvent (i.e.: methanol-water [15:85: v/v]) with known amounts of pure analyses. The matrix effects also were examined at three QC concentrations (2.5: 50: and 1250 nM for R- and S-warfarin; 0.5: 10: and 250 nM for S-7-OH-warfarin and (9R; 10S)-10-OH-warfarin) with quadruplicate samples at each concentration. Results were expressed as % signal remaining relative to standards in the reconstitution solvent: which represents100% signal remaining.
Freeze-thaw stability was evaluated by exposing QCs to three freezing (-80°C) and thawing (room temperature) cycles before sample preparation. The thermal stability was evaluated by comparing peak areas of standards in methanol-water (15:85: v/v) incubated at 50°Cfor 50 min (temperature and time needed during drying) to those incubated at 4°C for the same period of time. Sample stability in the auto sampler was evaluated by repeat-analysis of the same QCs stored in a thermostatic auto sampler (6°C) for 24 h.
The Limit of Detection (LOD) was defined as the lowest concentration of calibration standard that had a signal-to-noise ratio (S/N) greater than 10. The LLOQ was defined as 5 times the LOD with an accuracy of 80-120%: and an imprecision of <20%.
Analysis of human plasma samples from a clinical study
Blood samples were obtained from a healthy volunteer who participated in a clinical drug-drug interaction study involving warfarin as the victim drug. The human subject received placebo as the perpetrating drug and a single oral dose of 10 mg warfarin the morning of the study following an overnight fast. Venous blood (5 ml) was collected in K2-EDTAVacutainer® tubes (BD Biosciences: Franklin Lakes: NJ: USA) via an intravenous line at 0: 0.5: 1: 1.5: 2: 3: 4: 6: 8: 10: 12: 24: 36: 48: 60: 72: 96: 120: 144: and 168 h post-warfarin administration. Within 1 h following collection: blood samples were centrifuged (3000 rpm at 4°C for 10 min) and the resulting plasma samples transferred to pre-labeled cry tubes for storage at -80°C until analysis. The clinical study (Clinical Trials.gov registry number: NCT01250535) was approved by the Institutional Review Board of the University of North Carolina at Chapel Hill (Chapel Hill: NC: USA). The area under the plasma concentration–time curve (AUC): terminal elimination half-life (t1/2): maximum plasma drug concentration (C max): and time to reach C max (T max) were calculated using the trapezoidal rule–extrapolation method and non compartmental analysis (Phoenix Win Nonlin version 6.3; Pharsight: Mountain View: CA: USA).
Results and Discussion
HPLC-MS/MS analysis and enantiomeric separation
Enantiomeric separation of warfarin and warfarin-d5 (as IS) was achieved using a chiral HPLC column. The extracted ion chromatograms showed that both racemic warfarin and warfarin-d5 standards produced two well-resolved peaks at 4.44 and 4.80 min for warfarin (Figure 1A): and 4.43 and 4.78 min for warfarin-d5 (Figure 1B). These peaks are distinguishable from the hydroxylated warfarin metabolites due to their characteristic MRM transitions. Using synthetic optically pure R- and S-warfarin standards: it was determined that R-war far in eluted at 4.44 min and S-war far in at 4.80 min (data not shown). Due to the stereo chemical similarity between war far in and war far in-d5 (deuterated phenyl ring; Figures 1A and 1B): it can be postulated that R-war far in-d5 eluted at 4.43 min and S-warfarin-d5 at 4.78 min. Subsequently: R-warfarin-d5served as the IS for quantification of R-war far in and (9R; 10S)-10-OH-warfarin: while S-warfarin-d5 was used as the IS for quantification of S-war far in and S-7-OH-war far in.
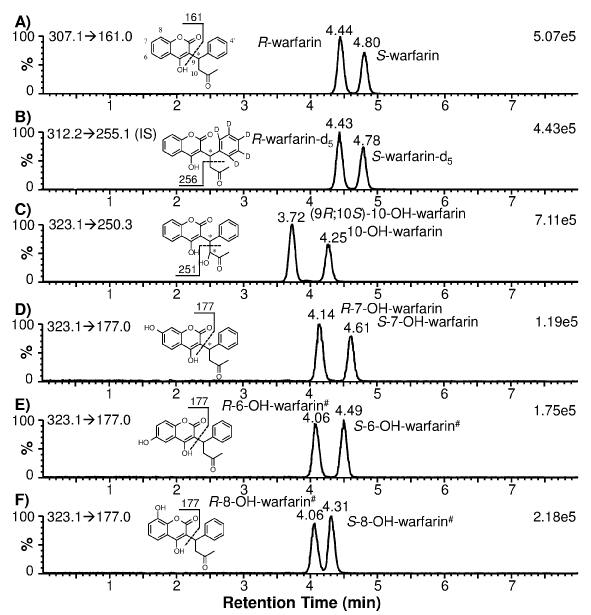
Figure 1: Extracted ion chromatograms of individual warfarin and hydroxylated warfarin standards (200 nM) prepared in blank human plasma. The monitored MRM transitions and signal intensity for the most intense peak are shown in the upper-left and upper-right corners of each chromatogram, respectively. The chemical structure of each analyte alsois shown with the postulated fragmentation and fragment mass. * denotes a chiral center; # denotes the postulated stereochemical assignment.
To determine CYP2C9 and CYP3A4 phenotypes: it is necessary to measure the formation of S-7-OH-warfarin (and/or S-6-OH-warfarin) and (9R; 10S)-10-OH-warfarin: respectively. Since 10-OH-warfarin can be distinguished from the hydroxylated benzopyran metabolites (i.e.: 6-: 7-: and 8-OH-warfarin) via its characteristic MRM transition (m/z 323.1→250.3): chromatographic separation of these hydroxylation metabolites is not necessary for HPLC-MS/ MS quantification of 10-OH-warfarin. However: 10-OH-warfarin contains two chiral centers (6 1c): This result in four possible stereoisomers. The stereochemistry of the10-OH-warfarin standard used in this study was not available from the vendor. Upon chiral separation and HPLC-MS/MS analysis: the 10-OH-warfarin standard produced two well-resolved peaks at 3.72 and 4.25 min (6 1C) or at 3.39 and 3.95 min (6 3A) using a same column of a different batch: corresponding to two of the four possible stereoisomers. It is known that CYP3A4 preferentially catalyzes the formation of (9R; 10S)-10-OH-warfarin from R-warfarin and (9S; 10R)-10-OH-warfarin from S-warfarin [6,7]. When R-warfarin was incubated with recombinant human CYP3A4: the predominant 10-hydroxylation metabolite co-eluted with the 3.39-min peak: confirming the identity of the 3.39-min peak as (9R; 10S)-10-OH-warfarin (6 3B). When S-warfarin was incubated with CYP3A4: the predominant 10-hydroxylation metabolite co-eluted with the 3.95-min peak: confirming the identity of the 3.95-min peak as (9S; 10R)-10-OH-warfarin (Figure 3B): the Enantiomeric stereoisomer of (9R; 10S)- 10-OH-warfarin. This analysis suggests that the 10-OH-warfarin standard used in this study was a racemic mixture of (9R; 10S)-10- OH-warfarin and (9S; 10R)-10-OH-warfarin. Hence it is reasonable to assume they had equal concentration in the racemic mixture: which allowed us to quantify (9R; 10S)-10-OH-warfarin in plasma samples (Figure 3C).
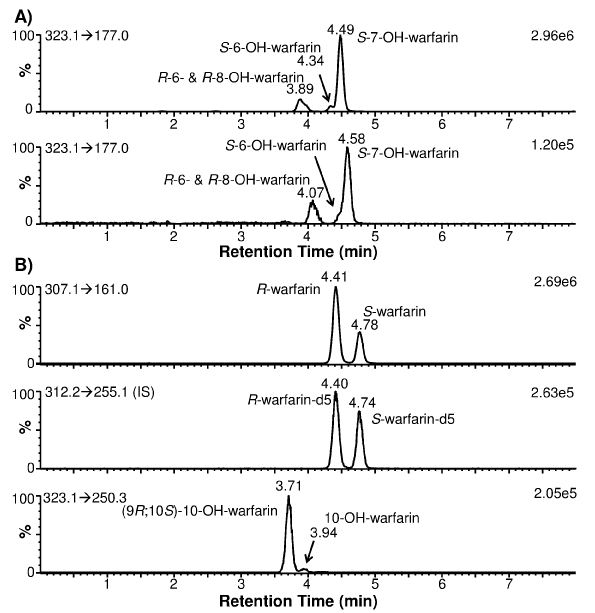
Figure 2: Extracted ion chromatograms of warfarin and its hydroxylation metabolites present in a human subject plasma sample obtained 36 h postwarfarin administration. (A) Chiral separation of S-7- from S-6-OH-warfarin was dependent upon the batch quality of the chiral HPLC column used. (B) (9R; 10S)-10-OH-warfarin was the predominant 10-OH-warfarin metabolite. The monitored MRM transitions and signal intensity for the most intense peak are shown in the upper-left and upper-right corners of each chromatogram, respectively.
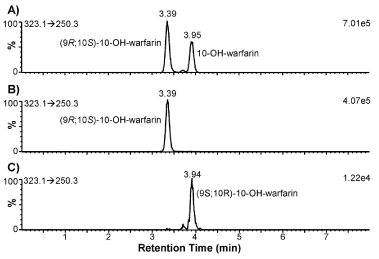
Figure 3: Extracted ion chromatograms of 10-OH-warfarin and its hydroxylation metabolites after 30 min incubation with recombinant human CYP3A4. (A) 10-OH-warfarin standard. (B) R-warfarin was used as substrate. (C) S-warfarin was used as substrate. The monitored MRM transitions and signal intensity for the most intense peak are shown in the upper-left and upper-right corners of each chromatogram, respectively.
To ensure specificity: enantiomeric separation of S-7-and S-6- OH-warfarin from the other hydroxylated benzopyran metabolites (i.e.: R-7-OH-warfarin: R-6-OH-warfarin: and 8-OH-warfarin) should be demonstrated because all of these metabolites have the same MRM transition (m/z 323.1→177.0): thus making them indistinguishable by MS/MS analysis alone. The extracted ion chromatograms for individual standards of 7-: 6-: and 8-OH-warfarin is shown in Figures 1D?F: demonstrating that the Enantiomeric pairs of these hydroxylated benzopyranmetabolites were separated from each other. The 7-OH-warfarin standard eluted at 4.14 and 4.61 min (Figure 1D). To determine which peak corresponded to the S-7-OH-warfarin metabolite: S-warfarin was incubated with recombinant human CYP2C9: as CYP2C9 preferentially catalyzes the formation of S-7-OH-warfarin [23]. Results indicated that S-7-OH-warfarin eluted at 4.61 min and R-7-OH-warfarin at 4.14 min (data not shown). Based on the observed elution order of warfarin and 7-OH-warfarin enantiomers (i.e.: R proceeds S): it can be postulated that R-6- and R-8-OH-warfarin co-elute at 4.06 min: while S-6- and S-8- OH-warfarin elute at 4.49 and 4.31 min: respectively (Figure 1E, 1F). Due to minimal HPLC separation of the hydroxylated benzopyran metabolites of R-warfarin: they are expected to co-elute if present in human plasma samples when analyzed using our method. Using a longer HPLC gradient (18 min): Zuo et al. [16] achieved a partial separation of these R-warfarin metabolites. However: since quantification of these metabolites was not the goal of our study: separation using the longer HPLC gradient was not attempted. The hydroxylated benzopyran metabolites of S-warfarin (i.e.: S-6-: S-7- and S-8-OH-warfarin) eluted at 4.49: 4.61: and 4.31 min (Figures 1D-F). However: S-6- and S-7-OH-warfarin were only partially separated. The extent of separation was found to be dependent upon the batch quality of the chiral HPLC column used (Figure 2A). As such: the chiral column with the best separation was used to analyze clinical plasma samples so that S-7-OH-warfarin could be specifically quantified.
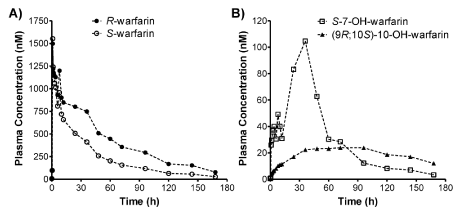
Figure 4: Plasma concentration-time profiles of R- and S-warfarin (A), as well as S-7-OH-warfarin and (9R; 10S) -10-OH-warfarin (B), for a human subject that received 10 mg of warfarin orally.
Calibration standards: accuracy and precision
Warfarin and 7-OH-warfarin used to prepare calibration standards were racemic mixtures of R and S stereoisomers: and 10-OH-warfarin has been shown to be a racemic mixture of (9R; 10S)-10-OH-warfarin and (9S; 10R)-10-OH-warfarin. Calibration standards for R-warfarin: S-warfarin: S-7-OH-warfarin and (9R; 10S)- 10-OH-warfarin were prepared by spiking blank human plasma with working standard mixtures. These calibration standards were then processed in a similar manner as clinical plasma samples: described in Section 2.4 “Sample preparation and HPLC-MS/MS analysis” under Section 2 “Materials and Methods”. The calibration range: LOD: S/N at LOD: representative calibration curves: and coefficient of determination is summarized in Table 1. The LLOQs obtained by this PPE-based method (using only 50 μl plasma) were much lower (~10 to 30-fold) than those using an SPE-based HPLC-MS/MS quantification method(using 200 μL plasma) [16] (Table 2): largely due to the more sensitive triple quadrupole MS instrument used in the current study. The enhanced assay sensitivity and calibration range will be essential for studies with limited sample availability (e.g.: capillary micro sampling [24]).
![]()
Compound
Calibration Rangea
S/N at LOD
Calibration Curveb
COD (R2)b
R-warfarin
0.25–5000 nM
=13
-0.0000139022�X2 + 0.562725�X-0.0504177
0.99949
S-warfarin
0.25–5000 nM
=12
-0.0000138943�X2 + 0.564179�X-0.0584107
0.99955
S-7-OH-warfarin
0.1–1000 nM
=5.4
0.0000258637�X2 + 0.258581�X-0.00957948
0.99967
(9R;10S)-10-OH-warfarin
0.1–1000 nM
=18
0.000118394�X2 + 0.6412�X-0.0203311
0.99972
Table 1: Calibration standards for warfarin enantiomers, S-7-OH-warfarin, and (9R; 10S)-10-OH-warfarin.
![]()
Reported Assays
Analytes Quantified
Extraction
(% Recovery)
Analysis Time
Dynamic Range
LLOQ
Zuo et al., 2010 [16]
warfarin enantiomers, stereoisomers of 7-OH-warfarin, and monohydroxylated warfarin metabolites� (4-, 6-, 8-, and 10-OH-warfarins)
mixed-mode cation-exchange cartridge
(> 87%)17 min
16.3–4890 nM (5–1500 ng/mL) for warfarin enantiomers
244–586 nM (75–180 ng/mL) for stereoisomers of 7-OH-warfarin16.3 nM (5 ng/mL) for warfarin enantiomers and stereoisomers of 7-OH-warfarin
0.2 mL of plasmaJones et al., 2011 [18]
warfarin enantiomers and monohydroxylated warfarin metabolites (4-, 6-, 7-, 8-, and 10-OH-warfarins)
PP by cold acetonitrile with 0.2% formic acid
(recovery not reported)17 min
2–1000 nM for all analytes
10 nM for all analytes
0.05 mL of plasmaWu et al. (reported herein)
warfarin enantiomers, stereoisomers of 7- and 10-OH-warfarin
PP by methanol-water [7:1, v/v]
(83–97%)6 min
0.25–5000 nM for warfarin enantiomers
0.1–1000 nM for S-7- and (9R;10S)-10-OH-warfarin1.25 nM for warfarin enantiomers�
0.5 nM for S-7- and (9R;10S)-10-OH-warfarin
0.05 mL of plasma
Table 2: Comparison of chiral LC-MS/MS analytical assays for warfarin and its hydroxylated metabolites formed by CYP2C9 and CYP3A4.
The intra-day accuracy and precision (CV) of QC samples at low: medium and high concentrations were 87.0 to 100.5% and 0.7 to 6.0% (Table 3). The inter-day accuracy and precision of QC samples were 92.3to 99.5% and 0.4 to 4.9% (Table 3). These values are well within the criteria of 15% bias and 15% CV provided in the US FDA guidance for bio analytical method validation [25].
![]()
Compound
QC Concentration (nM)
Intra-day
Inter-day
Accuracy (%)
Precision (%)
Accuracy (%)
Precision (%)
R-Warfarin
2.5
94.8
2.2
93.9
0.7
50
99.2
2.2
97.2
1.8
1250
98.5
1.0
98.8
1.1
S-Warfarin
2.5
95.1
1.4
94.1
1.0
50
99.7
2.3
98.1
2.0
1250
98.9
0.9
99.4
0.4
S-7-OH-warfarin
0.5
87.0
6.0
92.9
4.5
10
89.6
1.4
92.3
2.0
250
100.5
1.6
99.5
0.9
(9R;10S)-10-OH-warfarin
0.5
93.2
3.8
96
2.9
10
93.3
1.4
96.8
2.9
250
92.3
0.7
99.2
4.9
Table 3: Intra- and inter-day accuracy and precision for warfarin enantiomers, S-7-OH-warfarin, and (9R;10S)-10-OH-warfarin.
Extraction recovery: matrix effect and stability
One goal of this study was to simplify the warfarin extraction procedure: preferably avoiding SPE. PPE extraction recoveries of the four analyses in QC plasma samples are summarized in Table 3.They ranged from 82.9 to 95.6% and demonstrated reproducibility (SD < 8.8%). The recovery achieved with PPE is similar to or greater than that obtained with SPE (>80%) [16] or LLE (>73.8%) [14]. Minimal matrix effects were observed for R-warfarin: S-warfarin and S-7- OH-warfarin (Table 4): which all eluted later than (9R; 10S)-10- OH-warfarin (Figure 1). Significant matrix effects were observed for (9R; 10S)-10-OH-warfarin (~50% signal suppression; Table 4): but the matrix effects were quite reproducible (CV < 5.5%). Thus: it was important to prepare calibration standards in blank human plasma to mimic the matrix effects of human plasma samples.
![]()
Compound
QC Concentration (nM)
PPE Extraction
Matrix Effect
Recovery (%)
SD
(%)
Remaining Signal (%)
SD
(%)
R-Warfarin
2.5
94.4
6.7
96.7
3.4
50
86.7
3.6
95.2
2.1
1250
90.2
0.7
96.5
1.0
S-Warfarin
2.5
91.8
7.4
98.8
2.6
50
86.6
2.9
100.6
2.3
1250
94.4
5.5
100.5
0.7
S-7-OH-warfarin
0.5
87.7
4.9
89.8
5.0
10
84.1
1.9
83.5
3.3
250
87.5
6.9
81.4
1.2
(9R;10S)-10-OH-warfarin
0.5
82.9
8.8
49.2
2.7
10
88.2
3.3
52.2
1.6
250
95.6
2.1
50.4
0.7
Table 4: Extraction recovery and matrix effect for warfarin enantiomers, S-7-OH-warfarin, and (9R; 10S)-10-OH-warfarin.
The thermal stability of warfarin and its hydroxylated metabolites were evaluated at 50°C for 50 min: which are the temperature and time required to dry down PPE-extracted plasma samples. All four analyses were determined to be thermally stable with the percent remaining ranging from 93.7 to 101.4% (Table 5). The analyses also were stable after three freeze-thaw cycles with the percent remaining ranging from 96.9 to 113% (Table 5).
![]()
Compound
QC Concentration (nM)
Thermal Stability
Freeze-Thaw Stability
Remaining (%)
SD
(%)
Remaining (%)
SD
(%)
R-Warfarin
2.5
98.0
3.4
100.9
1.3
50
100.6
1.7
102.9
1.3
1250
98.4
1.6
101.9
2.5
S-Warfarin
2.5
98.6
3.2
100.5
1.2
50
101.4
0.9
101.6
1.2
1250
98.7
1.5
100.8
2.8
S-7-OH-warfarin
0.5
99.6
6.4
113.0
11.2
10
93.7
7.4
105.9
4.0
250
97.6
1.2
112.9
4.2
(9R;10S)-10-OH-warfarin
0.5
97.1
3.0
99.0
8.2
10
100.0
1.6
102.2
1.3
250
98.2
2.2
96.9
2.9
Table 5: Thermal and freeze-thaw stability for warfarin enantiomers, S-7-OH-warfarin, and (9R; 10S)-10-OH-warfarin.
Application to human plasma sample analyses
The validated PPE-based chiral HPLC–MS/MS method was applied for simultaneous quantification of R-warfarin: S-warfarin: S-7-OH-warfarin: and (9R; 10S)-10-OH-warfarin in human plasma samples obtained from a clinical drug-drug interaction study involving healthy human subjects. Representative extracted ion chromatograms of warfarin: warfarin-d5: 7-OH-warfarin: and 10-OH-warfarin for a plasma sample collected 36 h post-warfarin administration is shown in Figure 2B. The plasma concentration-time profiles of R-warfarin: S-warfarin: S-7-OH-warfarin: and (9R;10S)-10-OH-warfarin were plotted for a human subject that received 10 mg warfarin orally the morning of the study and was monitored for 7 days post-warfarin administration (Figure 4A, 4B). Pharmacokinetic measurements are summarized in Table 6. These values are in agreement with those previously reported for warfarin enantiomers [26].
![]()
Outcomes
Units
R-warfarin
S-warfarin
S-7-OH-warfarin
(9R;10S)-10-OH-warfarin
Cmax
nM
1,497
1,552
105
24
Tmax
h
1
1
36
72
AUClast
nM�h
68,800
39,500
5,100
3,180
AUC0→8
nM�h
73,800
40,800
5,270
4,470
t1/2
h
44
35
35
75
Table 6: Pharmacokinetics of warfarin enantiomers, S-7-OH-warfarin and (9R; 10S)-10-OH-warfarin in a human subject.
Conclusion
A simple PPE-based: highly sensitivechiral HPLC–MS/MS analytical method with short retention time has been developed and validated for simultaneous Enantiomeric quantification of R-warfarin: S-warfarin: S-7-OH-warfarin: and (9R; 10S)-10-OH-warfarin in human plasma. This method is expected to be more time-and cost-effective than SPE- or LLE-based methods. Furthermore: the outstanding sensitivity afforded by the combined high extraction recovery: low matrix suppression effect: and sensitive triple quadruple MS instrument will enable studies with limited sample availability (e.g.: capillary micro sampling). Specific quantification of S-7-OH-warfarin and (9R; 10S)-10-OH-warfarin will enable phenotypic characterization of CYP2C9 and CYP3A4 activities for drug-drug and drug-diet interaction studies involving these two CYP enzymes.
Acknowledgement
This work was supported by the National Institutes of Health research grant R01-GM089994 (MZW). MRS was supported by a training grant T32GM086330 from the National Institute of General Medical Sciences.
References
- Holbrook AM, Pereira JA, Labiris R, McDonald H, Douketis JD, Crowther M, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005; 165: 1095-1106.
- Whitlon DS, Sadowski JA, Suttie JW. Mechanism of coumarin action: significance of vitamin K epoxide reductase inhibition. Biochemistry. 1978; 17: 1371-1377.
- Verheugt FW. How can the bleeding risk associated with warfarin therapy be reduced? Nat Clin Pract Cardiovasc Med. 2008; 5: 14-15.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med. 2007; 167: 1414-1419.
- O'Reilly RA. Studies on the optical enantiomorphs of warfarin in man. Clin Pharmacol Ther. 1974; 16: 348-354.
- Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997; 73: 67-74.
- Lawrence RF, Rettie AE, Eddy AC, Trager WF. Chemical synthesis, absolute configuration, and stereochemistry of formation of 10-hydroxywarfarin: a major oxidative metabolite of (+)-(R)-warfarin from hepatic microsomal preparations. Chirality. 1990; 2: 96-105.
- Henne KR, Gaedigk A, Gupta G, Leeder JS, Rettie AE. Chiral phase analysis of warfarin enantiomers in patient plasma in relation to CYP2C9 genotype. J Chromatogr B Biomed Sci Appl. 1998; 710: 143-148.
- Osman A, Arbring K, Lindahl TL. A new high-performance liquid chromatographic method for determination of warfarin enantiomers. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2005; 826: 75-80.
- Ring PR, Bostick JM. Validation of a method for the determination of (R)-warfarin and (S)-warfarin in human plasma using LC with UV detection. Journal of pharmaceutical and biomedical analysis. 2000; 22: 573-581.
- Locatelli I, Kmetec V, Mrhar A, Grabnar I. Determination of warfarin enantiomers and hydroxylated metabolites in human blood plasma by liquid chromatography with achiral and chiral separation. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 2005; 818: 191-198.
- Naidong W, Lee JW. Development and validation of a high-performance liquid chromatographic method for the quantitation of warfarin enantiomers in human plasma. J Pharm Biomed Anal. 1993; 11: 785-792.
- Naidong W, Ring PR, Midtlien C, Jiang X. Development and validation of a sensitive and robust LC-tandem MS method for the analysis of warfarin enantiomers in human plasma. J Pharm Biomed Anal. 2001; 25: 219-226.
- Uno T, Niioka T, Hayakari M, Sugawara K, Tateishi T. Simultaneous determination of warfarin enantiomers and its metabolite in human plasma by column-switching high-performance liquid chromatography with chiral separation. Therapeutic drug monitoring. 2007; 29: 333-339.
- Takahashi H, Kashima T, Kimura S, Muramoto N, Nakahata H, Kubo S, et al. Determination of unbound warfarin enantiomers in human plasma and 7-hydroxywarfarin in human urine by chiral stationary-phase liquid chromatography with ultraviolet or fluorescence and on-line circular dichroism detection. Journal of chromatography B, Biomedical sciences and applications. 1997; 701: 71-80.
- Zuo Z, Wo SK, Lo CM, Zhou L, Cheng G, You JH. Simultaneous measurement of S-warfarin, R-warfarin, S-7-hydroxywarfarin and R-7-hydroxywarfarin in human plasma by liquid chromatography-tandem mass spectrometry. Journal of pharmaceutical and biomedical analysis. 2010; 52: 305-310.
- Zhou Q, Yau WP, Chan E. Enantioseparation of warfarin and its metabolites by capillary zone electrophoresis. Electrophoresis. 2003; 24: 2617-2626.
- Jones DR, Boysen G, Miller GP. Novel multi-mode ultra performance liquid chromatography-tandem mass spectrometry assay for profiling enantiomeric hydroxywarfarins and warfarin in human plasma. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2011; 879: 1056-1062.
- Kollroser M, Schober C. Determination of coumarin-type anticoagulants in human plasma by HPLC-electrospray ionization tandem mass spectrometry with an ion trap detector. Clin Chem. 2002; 48: 84-91.
- Ufer M, Kammerer B, Kirchheiner J, Rane A, Svensson JO. Determination of phenprocoumon, warfarin and their monohydroxylated metabolites in human plasma and urine by liquid chromatography-mass spectrometry after solid-phase extraction. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2004; 809: 217-226.
- Zhang ZY, King BM, Wong YN. Quantitative liquid chromatography/mass spectrometry/mass spectrometry warfarin assay for in vitro cytochrome P450 studies. Anal Biochem. 2001; 298: 40-49.