
Research Article
Austin J Anal Pharm Chem. 2016; 3(1): 1060.
Synthesis of Novel Chiral Phase Transfer Catalysts and Their Applications in Asymmetric Phase Transfer Catalytic Reactions
Zhang X, Wang Z, Xu P, Huang D, Dong X and Dai Z*
Department of Pharmaceutical Chemistry, School of Pharmacy, China Pharmaceutical University, China
*Corresponding author: Zhenya Dai, Department of Pharmaceutical Chemistry, School of Pharmacy, China Pharmaceutical University, P. R. China
Received: March 01, 2016; Accepted: March 22, 2016; Published: March 25, 2016
Abstract
Herein a serial of novel chiral phase transfer catalysts derived from cinchona alkaloids, containing a 8-member cycle, have been synthesized. The serial of catalysts have been applied to the asymmetric alkylation reaction and the asymmetric Darzens Reaction with a serial of satisfying and interesting results including moderate to excellent ee values (63-99.7%) in the asymmetric alkylation reactions and high diastereos selection and low to moderate ee values in the asymmetric Darzens Reaction (2.23:1 to trans only). An interesting phenomenon of asymmetric Darzens Reaction has also been discovered and explored, which probably concerning to the conjugation effect. A novel reasonable mechanism has also been put forward to explain this interesting phenomenon.
Keywords: Chiral phase transfer catalysts; Cinchona alkaloid; Asymmetric alkylation; Darzens reaction; Conjugative effect; Novel probable mechanism
Introduction
The development of chiral phase transfer catalysts derived from cinchona alkaloids is making its sense in economical, environmental and scientific fields [1,2]. The possible mechanism of asymmetric alkylation of glycine Schiff base was shown in Figure 1. The catalysts could transfer between the organic phase and the aqueous phase. In the aqueous phase, the anion of the catalysts Q+X- could be exchanged with the base MOH and the new organic base Q+OH- was obtained. The organic base Q+OH- was then transferred to the organic phase and the active α-H of the glycine Schiff base was deprived. The anion of the Schiff Base thus formed the chiral ion-pair together with Q+, then the asymmetric alkylation was realized with desired product formed and the catalysts Q+X- was reformed and re-transferred to the aqueous layer.
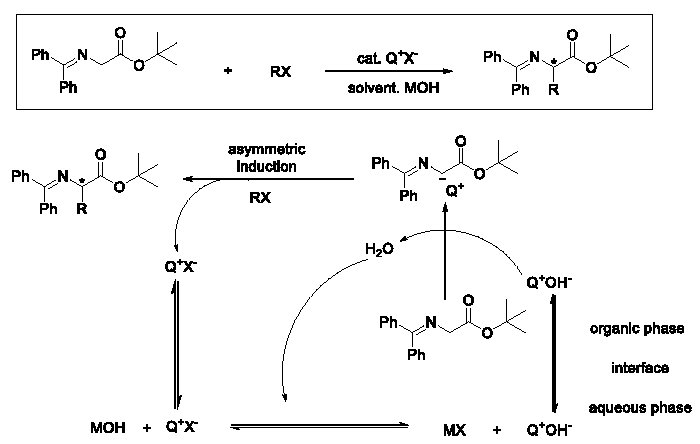
Figure 1: The possible mechanism of asymmetric alkylation of glycine Schiff
base.
Until recently, there have been three main generations of these catalysts derived from cinchona alkaloids (Figure 2). The first generation: R=H, Ar=Phenyl; the second generation: R=Allyl, Ar=Phenyl; and the third generation: R=Alkyl, Ar=Anthracyl. The first generation of catalysts were developed by Dolling’s group in 1984 [3,4], which were successfully applied in the asymmetric alkylation of glycine Schiff base by O’Donnell’s group with good enantioselectivity [5,6]. Deng et al. reported that the second generation of the catalysts could catalyze the asymmetric Darzens reaction with high yield and excellent enantioselectivity [7]. The third generation of the catalysts was developed by E.J. Corey’s group [8]. Recently Waser et al. reviewed the catalyzed asymmetric reactions catalyzed by the bifunctional ammonium catalysts [9], and Maruoka et al. also reviewed the asymmetric phase transfer catalysis with chiral quaternary ammonium catalysts derived from cinchona and chiral C2-type quaternary ammonium catalysts [10].
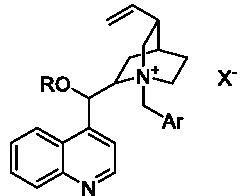
Figure 2: Cinchona alkaloids.
According to the theory stated by E. J. Corey, a tetrahedron was constituted by the nitrogen positive center of ammonium chiral phase transfer catalysts and four groups linked with it. The four groups should screen three faces of this tetrahedron, while the fourth face should be retained to accept the attack of the substrate. There should also be a nearby binding surface for attractive van der Waal’s interaction [8].
Result and Discussion
The cinchona alkaloids were essential for their enantioselectivity. In the synthesis of the cinchona alkaloids based chiral phase transfer catalysts, the hydroxyl and the bridgehead nitrogen were two key groups. Till now the modification of these two groups were processed separately. The method to modify them at the same time has not been reported yet. We wish to firstly report a serial of novel cinchona alkaloids based catalysts, which were modified these two groups at the same time. In these specific catalysts, a circular structure containing nitrogen and oxygen was brought in and played an important role in the steric screening to. At the same time, the cycle especially large cycle could also be flexible in the catalysis of the reactions. Considering to the large steric hindrance, we gave up the 6-member circular structure designation. Instead, the structure with 8-member cycle was attempted to be synthesized by cinchona alkaloids and 1,4-dibromobutane in Figure 3. On the other hand, the larger 8-member cycle could be more flexible than However, it was unavailable due to the poor activity of 1, 4-dibromobutane.
Figure 3: Cinchona alkaloids and 1,4-dibromobutane.
To improve the activity of the substrate, 1, 4-dibromobut- 2-ene was chosen to replace 1, 4-dibromobutane. Comparing to the commercial unavailable of (Z)-1, 4-dibromobut-2-ene, (E)-1, 4-dibromobut-2-ene was found to be commercially available. So we designed and synthesized the catalysts as shown in Figure 4.
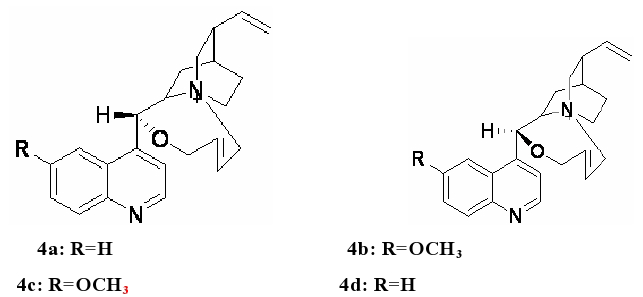
Figure 4: Design and synthesis of catalysts.
As was shown in Figure 4, these novel catalysts have a 8-member cycle. This cycle not only could screen the bridgehead nitrogen well, but also could be more flexible than smaller cycle such as 6-member or 7-member cycle. With these potential advantages, these catalysts should have excellent enantioselectivity and high ee value.
These novel catalysts could be synthesized as Figure 5. Cinchona alkaloids and (E)-1, 4-dibromobut-2-ene was heated to reflux with the base sodium hydride in the solvent of THF, with the yield of 60% to 70%.
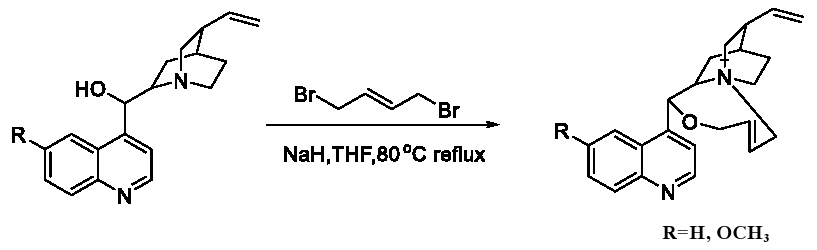
Figure 5: Synthesis of navel catalysts.
With these catalysts in hand, a serial of asymmetric alkylation were catalyzed by the novel catalysts. Firstly, the asymmetric benzylation was chosen as a model and several different conditions were investigated and the results were listed in Table 1.
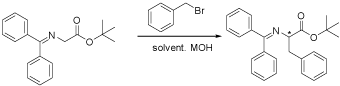
scheme:
![]()
.
Catalyst
Temperature
Base
Solvent
Yield(%)b
ee(%)c
Configurationd
1
4a
10°C
50%KOH
Toluene
87
99.7
S
2
4b
10°C
50%KOH
Toluene
75
80.1
S
3
4c
10°C
50%KOH
Toluene
77
17.8
R
4
4d
10°C
50%KOH
Toluene
80
60.7
R
5
4a
4°C
50%KOH
Toluene
60
88.4
S
6
4a
20°C
50%KOH
Toluene
71
90.1
S
7
4a
10°C
50%NaOH
Toluene
70
90.3
S
8
4a
10°C
50%CsOH
Toluene
74
91.2
S
9
4a
10°C
50%KOH
CH2Cl2
76
45.6
S
a. The reaction was carried out with 1.1 equiv. of benzyl bromide and 20.0 equiv. of 50% alkaline solution in the presence of 10 mol% 1-3 in different organic agents under the given conditions.
b. Isolated yields.
c. Enantiopurity was determined by HPLC analysis of benzylated imine using a chiral colume (DAICEl Chiralcel OD-H) with hexanes/i-PrOH as a solvent.
d. The absolute configuration was determined by comparison of the optical rotation with references [8,11].
Table 1: The asymmetric benzylation under different reaction conditions.
According to the results listed in Table 1, we found that catalyst 4a gave an excellent ee value while 4b to 4d gave poorer. Of all the bases, KOH gave the better ee while CsOH and NaOH gave poorer. Of all the temperature, 10oC was the best and of all the solvent, toluene was the best. So we chose 4a as the catalyst, 10oC as the temperature, KOH as the base and toluene as the solvent to catalyze several other asymmetric alkylation reactions with the results in Table 2.
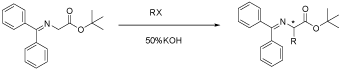
scheme:
![]()
RX
Yield(%)a
ee(%)b
Benzyl bromide
87
99.7
4-trifluoromethylbenzyl bromide
82
63
4-fluorobenzyl bromide
86
80
Iodomethane
77
71
Bromoethyne
75
87
a. Isolated yield
b. The enantiomeric excess was determined by HPLC analysis with a chiral column (DAICEl Chiralcel OD-H) with hexanes/i-PrOH as eluent.
Table 2: The asymmetric alkylation with different alkylation reagents.
According to the Table 2, we found that catalyst 4a could give high yield and excellent ee values in catalytic asymmetric alkylation. The highest ee was given by Benzyl bromide (99.7%).
After got the excellent results in asymmetric alkylation, the activity and enantio selectivity of these catalysts especially catalyst 4a were confirmed preliminary.
To further verify the activity and enantioselectivity of 4a, catalytic asymmetric Darzens reactions were processed. Until recently, there were several asymmetric Darzens reactions were reported. As was mentioned above, Deng’s group reported the asymmetric Darzens reactions using the second generation of the cinchona alkaloids based catalysts [7]. While Shioiri’s group reported diastereoselective Darzens reaction catalyzed by tetrahexylammonium bromide [12]. And high molecular phase transfer catalysts as Figure 6 applied in diastereoselective Darzens reaction was reported by Wang’s group [13]. Jonczyk’s group and Murugan’s group were also reported asymmetric Darzens reaction with different kinds of phase transfer catalysts [14,15].
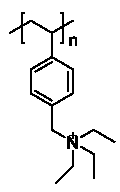
Figure 6: High molecular phase transfer catalysts.
Firstly, the Darzens reactions as Figure 7 were processed using the non-chiral catalyst TBAB (Table 3).
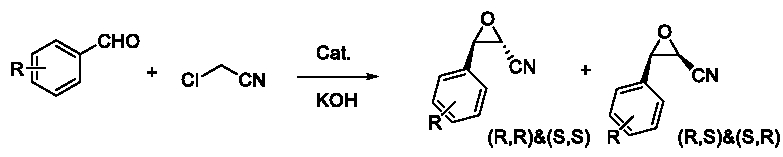
Figure 7: Darzens reactions were processed using the non-chiral catalyst
TBAB.
![]()
Entry
R
Catalyst
Yeilda
Time(h)
cis:trans
1
H
TBAB
70%
23
1.16:1
2
p-Cl
TBAB
13.30%
24
1.4:1
3
p-Br
TBAB
30%
12
1.4:1
4
p-Me
TBAB
19.50%
12
0.94:1
5
m-Me
TBAB
31.40%
12
0.77:1
a. Isolated yields including cis-product and trans-product.
`
Table 3: Darzens reactions catalyzed by TBAB.
In Table 3, we found that the rates of cis-product and transproduct were almost 1:1. Poor diastereos selectivity of the normal non-chiral phase transfer catalyst of TBAB was reflected in.
In continuation of our studies on the asymmetric phase transfer catalysis and Darzens reaction, we tried to investigate the novel chiral phase transfer catalysts 4a to 4d in the asymmetric Darzens reaction in Figure 7. Firstly, we processed the reaction with R= H in Figure 7 under different catalysts, solvent and base (Table 4).
![]()
Entry
Catalyst
Yeilda
Time(h)
Solvent
Base
cis:trans
ee of majorb
1
4a
66.9%
16
THF
KOH
2.23:1
70%
2
4b
60.1%
16
THF
KOH
2.05:1
61%
3
4c
55.7%
16
THF
KOH
1.5:1
45%
4
4d
58.6%
16
THF
KOH
1.3:1
53%
5
4a
--
--
toluene
KOH
--
--
6
4a
--
--
toluene
KOH(50% )
--
--
7
4a
50%
20
THF
NaOH
1.45:1
64%
8
4a
53%
18
THF
CsOH
1.37:1
58%
a. Isolated yields including cis-product and trans-product.
b. Enantiopurity was determined by HPLC analysis of benzylated imine using a chiral colume (DAICEl Chiralcel OD-H) with hexanes/i-PrOH as a solvent.
Table 4: Asymmetric Darzens reactions catalyzed by chiral PTCs.
In Table 4, we found that 4a could give the best result both in cis/ trans value and ee value. The best solvent and base were THF and KOH. The reaction could not be processed in toluene no matter the solid or aqueous solution of KOH was chosen.
After the best catalyst, base and solvent was chosen; we processed the reaction in Figure 7 with THF as the solvent, KOH as the base and 4a as the catalyst. The results were listed in Table 5.
![]()
Entry
R
Catalyst
Yeilda
Time(h)
cis:trans
ee of majorb
1
H
4a
66.90%
16
2.23:1
70%
2
p-Cl
4a
62.80%
9
3.8:1
35%
3
p-Br
4a
65.50%
8
4.1:1
50%
4
p-Me
4a
68%
8
5.8:1
17%
5
m-Me
4a
55.30%
8
6.9:1
30%
a. Isolated yields including cis-product and trans-product.
b. Enantiopurity was determined by HPLC analysis of benzylated imine using a chiral colume (DAICEl Chiralcel OD-H) with hexanes/ i-PrOH as a solvent.
Table 5: Asymmetric Darzens reactions of different aldehydes.
Comparing the results listed in Table 5 with Table 3, the catalyst 4a could give much higher yield than the common catalysts such as TBAB. Comparing to the long time of the reagents catalyzed by TBAB, 4a could push the reaction much faster. While in these reactions, quite excellent diastereoselectivity of 4a was shown when compared with TBAB, the common used phase transfer catalyst. The cis/trans rate ranges from 2.23 to 6.9 while the best of TBAB was just 1.4. The catalyst 4a could also show its limited enantioselectivity although it was not quite outstanding.
After that, we also applied 4a in a group of another Darzens reaction as Figure 8.
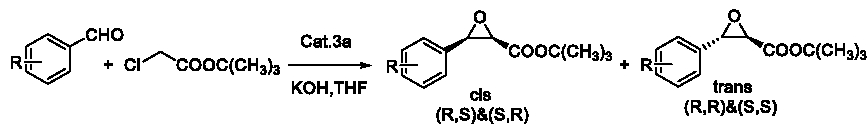
Figure 8: Darzens reaction.
This group of Darzens reactions were also be attempted to be catalyzed by TBAB and TEBAC. But these two catalysts could hardly catalyze these reactions in even 36h or 48h. But when the catalyst was replaced to 4a, these reactions could give an excellent and interesting result in Table 6, which was probably concerning to the conjugative effect of the substrate.
![]()
Entry
R
Catalyst
Yielda
cis:trans
1
H
4a
20%
1.5:1
2
p-Cl
4a
22.4%
1:4
3
p-Me
4a
27.8%
2:1
4
p-Br
4a
55.2%
1:1.4
5
p-NO2
4a
30%
Trans only
a. Isolated yields including cis-product and trans-product.
Table 6: Asymmetric Darzens reaction of different aldehydes.
According to Table 6, the selection of cis and trans expressed an interesting phenomenon that it seems to be concerned to the conjugative effect of the substrate. It seems that the rate of the transproduct depends on the density of the electron cloud on the aromatic ring which linked on the aldehydes group. When the density became lower and lower, the rate of trans-product would become higher and higher. When R= p-NO2, the density of the electron cloud became the lowest, so the product contains trans only. On the contrary, the higher density would bring the higher rate of the cis-product. However, we also found the research of the same reaction in Shioiri’s group and Jonczyk’s group reflected the same interesting phenomenon to our research [12,14].
To explain this interesting phenomenon, we put forward a probably mechanism of the cis-product. As was shown in Figure 9, this probably mechanism experiences 3 probably intermediate. Because of the potential stability of the envelop-shaped 5-member circular structure of the active intermediate 2, the intermediate 2 could be moment experienced from the known intermediate 1. So we could put forward our conjecture that these cis-products should experience the active intermediate 2.
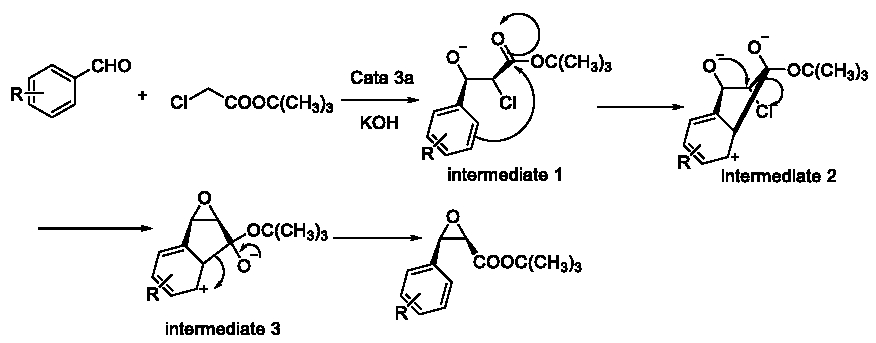
Figure 9: Mechanism of the cis-product.
With this potential mechanism in hand, we could easily explain the results in these reactions. When R= p-Cl, p-Br and p-NO2, the density of the electron cloud of the aromatic ring would become lower. Then there would be no plenty electron cloud to attack the positive center of the carbonyl group. So the intermediate 2 could not be arrived easily. Otherwise, the huge steric hindrance between Ph- and t-BuO- would prevent the cis-structure and push the major product trend to the more stable trans-product (Figure 10).
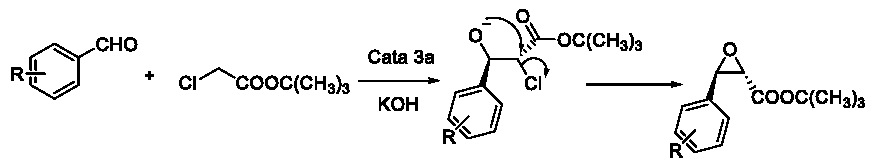
Figure 10: The huge steric hindrance between Ph- and t-BuO-.
While R=p-CH3, the density of electron cloud would become higher. Then the intermediate 2 would be arrived more easily, so the rate of the cis-product became higher. Otherwise, concerning to the high steric hindrance between Ph- and t-BuO-, the stability of the trans-product was still much higher than the cis-product. So according to Table 6, we could find that the rate of the cis-product could not increase obviously when R=p-CH3.
There was still one problem which could not be explained: why reactions in Table 5 could not obviously fit this potential mechanism? Otherwise, it probably could prove this potential mechanism on the other side. Comparing –CN to –COOC(CH3)3, we found that –CN has much lower steric hindrance but much higher electronwithdrawing effect. So firstly, the high electron-withdrawing effect of –CN could grip the electron cloud of the aromatic ring hugely, although when the density of the electron cloud was quite low, to arrive the intermediate 2 in Figure 11. While secondly, the low steric hindrance could guarantee the cis-structure of intermediate 2 to exist stably.
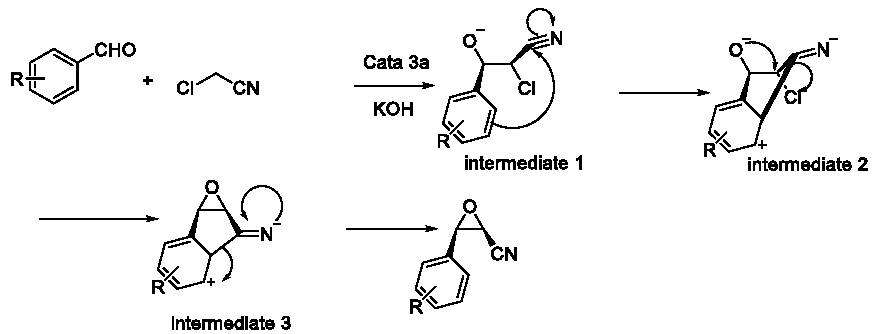
Figure 11: Cis-structure of intermediate 2.
This potential mechanism could explain the phenomenon in Table 4 and Table 6 easily. So we tried to verify this conjecture. We processed an extra experiment as Figure 12. Based on this conjecture, we predicted that the rate of cis-product should be higher than R= p-Cl and lower than R=H. The reason was that –Cl was an electron donating group in conjugative effect but an electron withdrawing group in inductive effect. When R= p-Cl, the active point of intermediate 1 became meta-position of the -Cl group. However, when R= m-Cl, the active point became para-position (Figure 13). Comparing p-Cl and m-Cl, the electron donating conjugative effect would guarantee the electron cloud density of p-position of the -Cl group higher than the m-position. But considering to the inductive effect, they were both lower than the density when R=H (Table 7).
Figure 12: Asymmetric Darzens reaction.
Figure 13: Asymmetric Darzens reactions.
![]()
Entry
R
Yielda
Time(h)
cis:trans
1
H
20%
12
1.5:1
2
m-Cl
52.5%
12
1:1
3
p-Cl
22.4%
12
1:4
a. Isolated yields including cis-product and trans-product.
Table 7: Asymmetric Darzens reactions of different aldehydes.
According to the result in Table 7, the result could preliminary confirm our conjecture about this mechanism. However, this mechanism was still just a reasonable conjecture till now. Further research and experiments would focus on the interesting phenomenon and the reasonable conjecture.
Experimental
Typical procedure of the synthesis of chiral catalyst 4a to 4d
(1S,7R,10S,E)-1-(quinolin-4-yl)-9-vinyl-1,3,6,8,9,10,11,11aoctahydro- 7,10-ethanopyrido[2,1-c][1,4]oxazocin-7-ium(4a): Cinchonidine (0.294g, 1mmol) was dissolved in THF (5ml), and sodium hydride (0.048g, 2mmol)was added. The reaction mixture was stirred and heated to 80 °C and refluxed for 1h, then (E) -1,4 - dibromo-2 - butane (0.321g, 1.5mmol) is added. The reaction mixture was further refluxed for 12 hours and the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated and the residue was purified with silica gel(chloroform: methanol = 30:1) to give the product (0.24g, 70% yield).
1H NMR(500 MHz, CDCl3): 8.9188-8.9043 (m, 1H, 8.2234-8.1656 (m, 2H), 7.7959-7.6351 (m, 2H), 7.4242-7.4097 (m, 1H), 6.4735- 6.4328 (m, 1H), 6.1463-6.0199 (m,1H), 5.8805 (s, 1H), 5.7594-5.6443 (m, 2H), 5.0114-4, 9213 (m, 3H), 4.8283-4.7940 (m, 1H), 3.4942- 3.4170 (m, 1H), 3.2820-3.1826 (m, 2H), 2.8837-2.7382 (m, 2H), 2.3971 (s, 1H), 1.9077-1.8730 (m, 3H), 1.6735-1.5299 (m, 2H)
13C NMR(500 MHz, CDCl3): 150.0278, 147.9949, 146.0408, 142.1767, 133.1039, 129.8867, 129.0374, 126.5786, 125.8792, 123.8068, 119.5861, 114.1071, 111.9291, 109.5361, 107.8249, 82.5908, 81.2770, 60.2081, 55.8686, 41.6474, 27.2226, 24.6763
ES-MS: 347.2 (M); [α]D 22 = +95.5°(c = 0.2 in CH2Cl2)
Elemental analysis: C: 79.5%, H: 7.8%, O: 4.6%, N: 8.1%
(1R,7R,10S,E)-1-(6-methoxyquinolin-4-yl)-9-vinyl- 1,3,6,8,9,10,11,11a-octahydro-7,10-ethanopyrido[2,1-c][1,4] oxazocin-7-ium(4b): Quinine (0.324g, 1mmol) was dissolved in THF (5ml), and sodium hydride (0.048g, 2mmol) was added. The reaction mixture was stirred and heated to 80 °C and refluxed for 1h, then (E) -1,4 - dibromo-2 - butane (0.321g, 1.5mmol) is added. The reaction mixture was further refluxed for 12 hours and the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated and the residue was purified with silica gel (chloroform: methanol = 30:1) to give the product (0.238g, 63% yield).
1H NMR(500 MHz, CDCl3): 8.7290-8.7204 (m,1H), 8.0506- 8.0238 (m,1H), 7.3945-7.3349 (m,2H), 7.2528-7.2218 (m,1H), 6.4336-6.4090 (m, 1H), 6.0792-6.0454 (m, 1H), 5.7119-5.6122 (m, 2H), 5.5391-5.5336 (m,1H), 4.9527-4.8838 (m, 2H), 4.7799-4.7595 (m, 1H), 3.9461-3.9340 (m, 3H), 3.3115 (s, 1H), 3.1652-3.0853 (m, 2H), 2.7290-2.6179 (m, 2H), 2.2770 (s, 1H), 2.1537 (s, 2H), 1.8751- 1.7688 (m, 4H), 1.5377-1.5175 (m, 3H), 1.2454-1.2100 (m, 1H)
13C NMR(500 MHz, CDCl3): 157.8505, 148.6980, 147.3576, 143.9983, 141.5296, 139.6108, 138.8185, 132.7581, 129.2598, 126.1965, 121.9891, 119.2893, 102.0956, 78.5066, 76.8134, 65.8451, 58.7737, 56.7155, 54.0146, 42.7227, 37.3716, 26.7743, 24.7174, 20.4527, 18.0087
ES-MS: 377.2 (M); [α]D 22 = +100°(c = 0.2 in CH2Cl2)
Elemental analysis: C: 76.4%, H: 7.7%, O: 8.5%, N: 7.4%
(1S,7R,10S,E)-1-(6-methoxyquinolin-4-yl)-9-vinyl- 1,3,6,8,9,10,11,11a-octahydro-7,10-ethanopyrido[2,1-c][1,4] oxazocin-7-ium(4c): Quinidine (0.324g, 1mmol) was dissolved in THF (5ml), and sodium hydride (0.048g, 2mmol) was added. The reaction mixture was stirred and heated to 80 °C and refluxed for 1h, then (E) -1,4 - dibromo-2 - butane (0.321g, 1.5mmol) is added. The reaction mixture was further refluxed for 12 hours and the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated and the residue was purified with silica gel (chloroform: methanol = 30:1) to give the product (0.2g, 53.1% yield).
1H NMR(500 MHz, CDCl3): 8.7204-8.7193 (m,1H), 8.0601- 8.0294 (m, 1H), 7.4040-7.3413 (m, 2H), 7.2626-7.2166 (m,1H), 6.4454-6.4046 (m, 1H), 6.1093-6.0400 (m, 2H), 5.6780-5.5472 (m, 2H), 5.1599-5.0716 (m, 2H), 4.9482-4.8920 (m, 1H), 4.7891-4.7547 (m, 1H), 3.9442-3.9328 (m, 3H), 3.1868-3.1019 (m, 2H), 2.9826- 2.7679 (m, 4H), 2.2706-2.2463 (m,1H), 1.7724 (s, 1H), 1.5069-1.4824 (m, 3H), 1.2527-1.1816 (m, 2H), 0.9454-0.8975 (m, 1H)
13C NMR(500 MHz, CDCl3): 157.1687, 150.1687, 147.3799, 145.8176, 144.0027, 140.8254, 133.1505, 131.2325, 129.6808, 126.9962, 121.2880, 119.2112, 102.2112, 82.2055, 80.6039, 60.2081, 55.4593, 49.9428, 49.0488, 48.3823, 39.6256, 27.7150, 25.9084, 24.6867
ES-MS: 377.2 (M); [α]D 22 = -14°(c = 0.2 in CH2Cl2)
Elemental analysis: C: 76.4%, H: 7.7%, O: 8.5%, N: 7.4%
(1R,7R,10S,E)-1-(quinolin-4-yl)-9-vinyl-1,3,6,8,9,10,11,11aoctahydro- 7,10-ethanopyrido[2,1-c][1,4]oxazocin-7-ium(4d): Cinchonine (0.294g, 1mmol) was dissolved in THF (5ml), and sodium hydride (0.048g, 2mmol) was added. The reaction mixture was stirred and heated to 80 °C and refluxed for 1h, then (E) -1,4 - dibromo-2 - butane (0.321g, 1.5mmol) is added. The reaction mixture was further refluxed for 12 hours and the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated and the residue was purified with silica gel (chloroform: methanol = 30:1) to give the product (0.197g, 56.9% yield).
1H NMR(500 MHz, CDCl3): 8.9185-8.9043 (m, 1H), 8.1907- 8.1148 (m, 2H), 7.7660-7.6210 (m,2H), 7.4260-7.4120 (m, 1H), 6.4651-6.4240 (m, 1H), 6.1454-6.0176 (m, 2H), 5.9080-5.8020 (m,1H), 5.7185-5.6410 (m, 1H), 5.1782-5.0697 (m, 2H), 4.9745- 4.9180 (m,1H), 4.8213-4.7874 (m,1H), 3.2700-3.1762 (m, 3H), 3.0660-2.9939 (m,2H), 2.8654 (s, 1H), 2.3483-2.1630 (m, 3H), 1.8380 (s, 2H), 0.9761-0.8701 (m, 2H)
13 NMR(500 MHz, CDCl3): 149.9942, 147.9244, 145.8366, 140.6015, 133.0530, 129.8156, 129.0673, 126.6098, 125.8520, 123.8260, 119.3837, 118.8478, 82.0510, 80.6292, 68.2785, 59.9805,49.0344, 48.1538, 27.6099, 25.6839, 24.5560, 23.5513
ES-MS: 347.2 (M); [α]D 22 = +6°(c = 0.2 in CH2Cl2)
Elemental analysis: C: 79.5%, H: 7.8%, O: 4.6%, N: 8.1%
Typical procedure of the asymmetric alkylation of glycine derivatives
To a mixture of tert-butyl 2-((diphenylmethylene)amino)acetate (0.148 g, 5 mmol), catalyst 4a (10% mol, 20 mg) in 5 ml toluene was added benzyl bromide (1.1eq), and KOH (20eq, 50% aqueous) at 10 °C. The reaction mixture was stirred vigorously at the same temperature for 18 hours. Then water (5 ml) was added, the organic layer was separated and the aqueous layer was further extracted with CH2Cl2 twice. The organic layer was combined, washed with brine, dried over Na2SO4. Evaporation of the solvent gave the crude product, which was purified with preparative TLC (PE:EA=50:1) to give the product (0.168g) as oil.
1H NMR(500 MHz, CDCl3) : 7.82-7.81(m, 2H), 7.38-7.28(m, 6H), 7.25-7.14(m, 3H), 7.08-7.06(m, 2H), 6.64-6.62(m, 2H), 4.15-4.11(m, 1H), 3.27-3.15(m, 2H), 1.45(s, 9H). [α]D 22 = -210°(c = 0.2 in 2Cl2). HPLC analysis retention time: 10.72min, 13.41 min (major) (Daicel Chiralcel OD-H, hexane/i-PrOH = 99.5/0.5, flow rate = 0.5 mL/min)
Typical procedure of the asymmetric Darzens reactions
To a mixture of 0.106g benzaldehyde (1mmol), 0.091g chloroacetonitrile (1.2mmol) and 5ml THF, 0.035g 4a (0.1mmol) was added and stirred for 20min. 0.067g KOH (1.2mmol) was added and continued stirring for 16h. The mixture was filtered and purified by TLC (PE:EA=50:1) to give the cis-product 0.067g and trans-product 0.03g as colorless oil [6].
Cis-product 1H NMR(500 MHz, CDCl3) : 7.408~7.388(3H,m), 7.282~7.263(2H,m), 4.278~4.275(1H, m), 3.410~3.405(1H,m)
Trans-product 1H NMR(500 MHz, CDCl3) : 7.245~7.260 (5H,m), 4.248~4.237 (1H,m), 3.778~3.766 (1H, m)
[α]D 22 =+41o (major product)
Conclusion
We successfully designed and synthesized a serial of new chiral phase-transfer catalysts with the structure of 8-atom cycle based on the cinchona alkaloid. These new catalysts were applied in the asymmetric alkylation of glycine derivatives with high yield and moderate to excellent enantioselectivity. When applied to asymmetric Darzens reactions, fast reaction rate, high yield and excellent diastereoselectivity & enantioselectivity were given. An interesting and unknown phenomenon was also observed and a reasonable mechanism was put forward to explain this phenomenon. Further work is under way to confirm the novel mechanism and improve the enantioselectivity and diastereoselectivity.
Acknowledgment
We were thankful to the National Natural Science Foundation of China for their financial support (No. 21102180).
References
- Ooi T, Maruoka K. Asymmetric organocatalysis of structurally well-defined chiral quaternary ammonium fluorides. Acc Chem Res. 2004; 37: 526-533.
- Maruoka K. Practical aspects of recent asymmetric phase-transfer catalysis. Org Process Res Dev. 2008; 12: 679-697.
- Dolling UH, Davis P, Grabowski EJJJ. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. Am. Chem. Soc.1984; 106: 446-447.
- Hughes DL, Dolling UH, Ryan KM, Schoenewaldt EF, Grabowski EJJJ. Efficient catalytic asymmetric alkylations. 3. A kinetic and mechanistic study of the enantioselective phase-transfer methylation of 6,7-dichloro-5-methoxy-2-phenyl-1-indanone. Org. Chem. 1987; 52: 4745-4752.
- O’Donnell MJ, Bennett WD, Wu SJ. The stereoselective synthesis of .alpha.-amino acids by phase-transfer catalysis. Am. Chem. Soc. 1989; 111: 2353-2355.
- Lipkowitz KB, Cavanaugh MW, Baker B, O’Donnell MJJ. Theoretical studies in molecular recognition: asymmetric induction of benzophenone imine ester enolates by the benzylcinchoninium ion. Org. Chem. 1991; 56: 5181-5192.
- Liu Y, Provencher BA, Bartleson KJ, Deng L. Highly Enantioselective Asymmetric Darzens Reactions with a Phase Transfer Catalyst. Chem Sci. 2011; 2: 1301-1304.
- Corey EJ, Xu F, Noe MCJ. A Rational Approach to Catalytic Enantioselective Enolate Alkylation Using a Structurally Rigidified and Defined Chiral Quaternary Ammonium Salt under Phase Transfer Conditions. Am. Chem. Soc. 1997; 119: 12414-12415.
- Novacek J, Waser M. Bifunctional Chiral Quaternary Ammonium Salt Catalysts: A Rapidly Emerging Class of Powerful Asymmetric Catalysts. Eur. J. Org. Chem. 2013; 4: 637-648.
- Hashimoto T, Maruoka K. Recent development and application of chiral phase-transfer catalysts. Chem Rev. 2007; 107: 5656-5682.
- Okino T, Takemoto Y. Asymmetric alkylation of tert-butyl glycinate Schiff base with chiral quaternary ammonium salt under micellar conditions. Org Lett. 2001; 3: 1515-1517.
- Arai S, Suzuki Y, Tokumaru K, & Shioiri T. Diastereoselective Darzens reactions of α-chloroesters, amides and nitriles with aromatic aldehydes under phase-transfer catalyzed conditions. Tetrahedron let. 2002; 43: 833-836.
- Wang, Zongting, et al. Efficient Darzens condensation reactions of aromatic aldehydes catalyzed by polystyrene-supported phase-transfer catalyst. Jour. Mole. Cat. A: Chem. 2004; 218.2, 157-160.
- Jonczyk A, Zomerfeld T. Convenient synthesis of t-butyl Z-3-substituted glycidates under conditions of phase-transfer catalysis. Tetrahedron Let. 2003; 44: 2359-2361.
- Murugan E, Siva A. Effective Darzen's condensation of 4-nonanolide with 1,6-dibromohexan-2-one using new bead-shaped insoluble multi-site (six-site) phase transfer catalyst and low concentration of aqueous NaOH. Jour. Mole. Cat. A: Chem. 2007; 277: 81-92.