
Review Article
Austin J Anal Pharm Chem. 2017; 4(1): 1078.
Implementing Quality by Design (QbD) in Chromatography
Thakor NS1* and Amrutkar SV2
¹Department of Pharmaceutical Chemistry, SSR College of Pharmacy, Sayli Silvassa, India
²Department of Pharmaceutical Chemistry, Sir Dr. M.S. Gosavi College of Pharmaceutical Education, Nashik, India
*Corresponding author: Thakor NS, Department of Pharmaceutical Chemistry, SSR College of Pharmacy, Sayli Silvassa, UT of DNH, India
Received: December 14, 2016; Accepted: January 05, 2017; Published: January 09, 2017
Abstract
Analytical methods are required to be developed at different stages of the pharmaceutical product life cycle. These activities if properly not streamlined based on the knowledge of science and process understanding may lead to a very costly and time consuming procedure. Pharmaceutical industries are striving for new policy and/or new element which can be added/replaced the existing elements of quality and risk management system. Quality by design (QbD) approach is one of the alternatives where systematic implementation of Quality Management programme can be established by better knowledge of the method parameters. Recently the concept has been also appreciated by different regulatory, especially by EMA (Europe Medicines Agency) and other ICH countries authorities over the globe. When using QbD concept for development of analytical method, analytical chemist in factstudy the additive and interactive effects of all method parameters on its continual performance. Further by using statistical mockups method operable design region (MODR) is determined. Changes within MODR are not significant and revalidation of the method is not required. Current article reviews how QbD approach can be more expressively used for chromatographic analytical method development for pharmaceuticals.
Keywords: QbD; Analytical method; Pharmaceuticals; Chromatography
Introduction
Analytical method development and validation plays a very crucial role in product development. A robust analytical method not only assures whether the quality of drug is achieved as per the intended therapeutic use but also serves as a purity check at each stage of product development life cycle. With the commercial manufacturing of product it is important that the analytical method is time saving as well as robust and accurate since the release in market is decided on final quality control results of finished product accompanied by other data of the batch. Analytical techniques widely include estimation of physical, chemical, physicochemical, and/or biological parameter of the substance of interest. Use of chromatographic analytical techniques such as High performance liquid chromatography (HPLC), Gas chromatography (GC), High performance thin layer chromatography (HPTLC), super critical fluid chromatography (SFC): are very widely known as they have various advantages over other non-chromatographic methods. They are versatile, robust, and require fewer amounts of samples. With the use of automation these techniques minimize the probability of human error.
The main concern of the analytical chemist is to develop a suitable analytical method that that exactly works as per the intended use. In the current scenario of analytical chemistry, there are 2 approaches followed for analytical method development. The former is based on trial and error which studies one factor at a time (OFAT) where one parameter alone is optimized for the expected response whilst others remained constant. This practice always yields a narrow robust behavior of the method for instrumental variables used in method development phase. Hence the strategy of analytical method (i.e., OFAT) development has high risk in method failure and always requires revalidation protocol after method transfer or during alternative method development; thereby it has been increasing the cost of the method.
The later approach is Analytical Quality by Design (AQbD) which explores scientific understanding in method implementation sequences and starts with product quality that relates the risk assessment in method choice and then between method parameter and expected method results and finally a region for high robust and cost effective approach. Design of Experiment (DoE) is a part of AQbD, and it represents the interaction among the input variables that ultimately affect the method response and results. AQbD paradigm is a preferred and recommended strategy to be followed in analytical method development so as to attain regulatory flexibility and reduce Out of specification (OOS), Out of term (OOT) and Out of control (OOC) results and when this approach is used to study any chromatographic method, the methods explores more knowledge of the parameters that has to be controlled and monitored during the life cycle of the method.
The scope of AQbD approach is briefly represented in Figure 1.
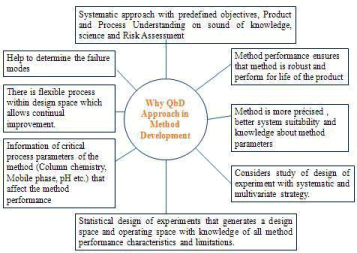
Figure 1: Scope of AQbD in Analytical method development.
AQbD and Chromatography
There a very less or no detailed literature published on how to implement AQbD approach in method life cycle. Nevertheless this approach is very widely known in development/optimization of pharmaceutical formulation or processes. Whenever an analyst thinks to apply DOE, he may have a misperception that the model uses more trials or practically it is difficult to select a design or interpret data statistically. However this is very untrue as these models have proven equally efficient if used in a proper way.
Related reference is provided in United States Pharmacopoeia (USP-NF) and European Pharmacopoeia (EP) where flexibility is granted for an analytical method that can be changed without the need for revalidation if AQbD approach has been implemented. However further guidelines are not given [1]. Some researchers have however compiled the data /utilized AQbD approach successfully for chromatographic methods [2-9].
In context with the approach proposed in the United States Food and Drug Administration (US FDA) guidelines for a three-stage approach could be taken with method validation.
A. Stage 1- Method Design: Define method requirements and conditions and identify critical controls.
B. Sage 2 -Method Qualification: Confirm that the method is capable of meeting its design.
C. Stage 3- Life cycle Management: Gain ongoing assurance to ensure that the method remains in a state of control during routine use (Figure 2).
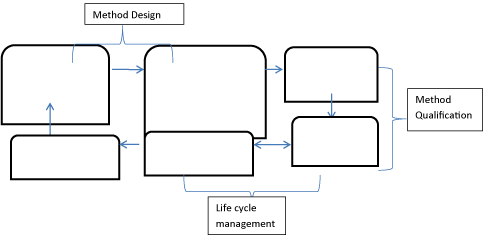
Figure 2: Schematic diagram of Method development life cycle as per AQbD.
A. Stage 1-Method design
Selecting/Designing an analytical method fit for the intended purpose are the first and foremost step in method lifecycle. Various elements while selecting an appropriate method of analysis should be given a due consideration. Some of the parameters include molecular properties (physical and chemical) of analyte, its related impurities, cost of reagents, sensitivity and selectivity of method, availability of instrument, time required for analysis. For a molecule with significant Ultra violet (UV) chromophore Liquid Chromatography (LC) with UV detector is appropriate method of detection but in case of no chromophore Gas Chromatography consideration may be given to other chromatographic methods or derivatization methods. In addition presence of ionizable centers, polarity of molecule should be reread.
Method design in terms of AQbD includes space generation, defining Analytical Target Profile (ATP), experimental design screening and establishing Critical Quality Attributes (CQA’s).
Define ATP: At this stage the analytical chemist defines and documents the objectives or performance requirements of the method. In other words ATP states method’s purpose which further initiates method selection, design and development activities. To construct the ATP, it is necessary to identify the characteristics of method that will be indicators of method performance. Once these characteristics are identified the next stage is to determine the acceptance criteria of the characteristics. Knowledge regarding the process variation and its impact on the quality is helpful in deciding the acceptance criteria.
Analytical methods can be designed and developed for many purposes such as for qualitative purpose, for stability study of active pharmaceutical ingredient (API) or formulation, impurity profiling, preservative content or to estimate analyte in the biological fluids. Each type of method will have its predefined purpose and target.
Typical examples of the ATP’s are summarized in Table 1.
![]()
Routine quantitative methods for API, finished products or intermediates.
Accurate quantitation of analyte without interference of matrix components
For stability indicating method
Accurate determination of degradants without interfering analyte under study
For impurity profiling method
Accurate determination and quantitation of related impurities of analyte
For Bioanalytical method
Accurate determination and quantitation of analyte in biological matrix
Table 1: Examples of ATP’s for analytical methods.
Chromatic methods whenever developed should be robust, simple and accurate method where the chromatograms are sharp and well resolved from each other. The method should be specific to the analyte and should have a least interference from matrix components or mobile phase.
Establish CQA’s: At this point the analyst has to identify the critical method parameters that directly influence the method performance. For this exercise a deep understanding of the method is required. This can be done by literature scouting or referring retrospective data of analyte to be determined. The CQA’s will differ from project to project. Critical method parameters (CMP’s) are divided into three types’ viz. parameters regarding analyte, parameters regarding instrument and parameters regarding operation conditions. Typical CMP’s for chromatographic experiments are sampling, sample preparation, standards, reagents, column chemistry, mobile phase composition, pH and flow of mobile phase, column temperature, detector selection etc. CQA’s (responses) for the above parameters would be resolution, retention time, tailing factor, detection limit, robustness etc. Never the less when we establish CQA’s we simply list out all the factors that affect the method performance.
Perform risk assessment: At this stage the analyst has to link CMP’s to CQA’s. Critical parameters of method are screened and their risk on methods performance is assessed. Analyst can use several risk assessment tools such as Fishbone diagram and Failure Mode Effect Analysis (FMEA’s). Both the tools when used identify the elements/ operations within the system that render it vulnerable. The output/results of these tools can be used as a basis for generating design space or further analysis or to guide resource deployment. Risk matrices are used to assess parameter risks with respect to the relevant attributes (e.g., accuracy, precision, resolution, tailing). At early developmental phase, simple matrices are built from scientific knowledge and experience of the methodology. As programs progress in development, the use of more detailed matrices (e.g., FMEAs, Cause and Effect (C&E) analysis (Figure 3) can facilitate the identification of high risk method parameters and attribute responses for subsequent experimentation to ensure that they do not impact the method’s ability to meet the ATP. This step shall help the analyst to understand deeply about the parameters that affect the methods performance. It will also give a preliminary knowledge about the design space of the method and the boundaries of method parameters which have to be controlled stringently to fulfill the intended purpose of the method. To perform risk assessment study of the proposed analytical methods a deep knowledge of physical and chemical properties of analyte is required. If the analyte has to be resolved with a mobile phase that require use of certain pH conditions the risk prone due to slight variation of pH should be studied. The risk that a method can face during its operational conditions should be listed out and ranked and all risks that directly impact the method should be documented which can be referred during method developmental stage.
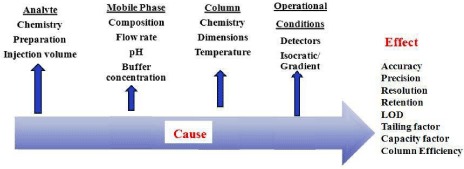
Figure 3: Cause effect diagram for risk assessment of Chromatographic
method by AQbD.
Generate design space: The term design space is well put forward in ICH Q8 [5] guideline, which states that it is “the multidimensional combination and interaction of input variables and process parameters that have been demonstrated to provide assurance of quality”. Design space can be thus a set of ranges for variables that have been proven to be effective. In chromatographic method when influence of all parameters and interaction of the same on method is evaluated by some statistical design the outcome would generate a design. This design shall be moreover combination of different ranges of mobile phase combination, pH, columns, flow rates etc. which gives results that are more close to the set objectives. Preliminary the trials can be conducted based on referencing, knowledge etc. where the analyst gets a very fair idea regarding a definite region of the method where deliberate changes do not affect methods performance. Movement within this design space is not considered as a change therefore design space supports increased flexibility of the method for regulatory approvals [6]. Thus design space is also termed as ‘method operable design region’ (MODR). It is this MODR which is then said to encompass all possible method condition combinations for which the predefined method objectives are met. The optimized method parameters are then said to lie somewhere around this region where they have to be optimized by carrying out a set of experiments. MODR hence provides the confidence that the area around the method conditions is robust and rugged. Various trials of different mobile phase combinations, columns, flow rates, column temperatures, pH of mobile phase etc. can be tried for preliminary optimization. The outcome of these trials should be documented and the best suitable combination can be further subjected to statistical evaluation. An example of summary of different trials is tabulated in Table 2 where how effect of changes in mobile phase combination and composition and variation in pH effects on retention time, theoretical plates, tailing factor and peak shape are compared. A combination which is most suitable for a good resolution will be selected for design of experiment.
![]()
Trial No.
Column
Mobile phase
pH
Retention time for analyte
Number of theoretical plates
Tailing factor
Results
1
C18
(Brand A)
Methanol : water
(80:20)
-
9.23
93
1.58
Broad peak
2
C18
(Brand A)
Acetonitrile :water (80:20)
-
17.53
341
1.12
Late elution
3
C18
(Brand A)
Methanol : phosphate buffer 0.01M
3.14
3.52
357
1.26
Splitting of peak
4
C18
(Brand A)
Acetonitrile
phosphate buffer 0.01M
3.14
3.09
400
1.72
Distorted peak
5
C18
(Brand A)
Acetonitrile
Methanol
phosphate buffer 0.01M
3.14
3.17
1500
2.35
Hump
6
C18
(Brand A)
Methanol: 0.4%TEA in water
3.0
4
1160
4
Broad peak
7
C18
(Brand B)
Methanol
Ammonium acetate buffer0.01M
5.01
12.48
300
1.66
Run time more
8
C18
(Brand B)
Methanol
Ammonium acetate buffer0.01M
3.80
3.58
5906
1.50
Satisfactory
*Data tabulated is only for understanding purpose and does not reveal any actual experiments performed.
Table 2: Preliminary system suitability Trials.
Using design of experiments for method screening and optimization: Design of experimental approach for method optimization is a powerful tool for analytical chemist. Some of the advantages include, factors and interactions are assessed with maximum efficiency, conclusions apply to a wide range of conditions since factor effects are measured at varying levels and maximum use is made of the data since all main effects and interactions are calculated from the data. Experimental designs are further classified into 4 types viz. Factorial designs and modifications, Central composite design, Mixture design and D-optimal design.
Factorial designs consist of a series of experiments in which factors are varied simultaneously, rather than one at a time. The most common statistical designs that are used to study interaction of input variables and response are full factorial and fractional factorial designs. Full Factorial design involves studying the Effect of all the factors (n) at various levels (x) including the interaction among them, with the total number of experiments as xn. They are often denoted in a matrix format known as the design matrix. The most basic factorial design examines each factor at two separate levels known as a twolevel factorial design, the number of experiments required can be determined by the formula n = 2x where n represents the number of experiments and x is the number of factors being studied. Therefore, a three level factorial design for optimizing two factors would require a total of 32 or 9 experiments.
Factorial designs are of 2 types symmetric where each factor has same number of levels (22 or 33) and asymmetric where the number of levels differs for each factor (23 or 32). Fractional factorial design is used to examine multiple factors efficiently with fewer runs than corresponding full factorial design. These are applied when two or more treatments do not interact, factorial designs can test the main effects of each using smaller sample sizes and greater precision than separate parallel group designs.
An application 32 of factorial design for 2 factors mobile phase combination and flow rate of best combination (trial no.8 indicated in Table 3), where 0 levels for flow rate is 1mL/min and mobile phase composition is 45:55 v/v respectively. Table 4 summarizes the number of trials for each set of combination of factor levels. Mobile phase composition with respect to methanol is varied and accordingly the composition of buffer to make 100mL is changed.
![]()
TABLECREATED
Table 3: Critical parameters and their levels.
![]()
TABLECREATED
Table 4: Experimental runs with different combinations.
Similarly all the runs are performed and the influence of the above three factors is evaluated on the responses that are shown in cause effect diagram. Results are analyzed by various software such as Minitab, SAS, SPSS or even Microsoft Excel, Fusion AE, Design Expert etc.
Most common experimental designs which have been applied to chromatography are full factorial, fractional factorial, Plackett –Burman and Box – Behnken design. General comparison and application of each design is summarized in Table 5.
![]()
TABLECREATED
Table 5: Comparative summary of various Factorial designs.
By making use of above software the outcome of 9 trials on system suitability parameter can be studied and the desirability plots can be obtained. Some software may also suggest in between combination like flow rate of 1.2min/mL or 1.4min/mL or mobile phase combination of 49.5:50.5 v/v can be additionally tried for best optimization w.r.t system suitability.
B. Stage 2-Method qualification
At this point the analytical chemists assure that the designed method operates as per requirement. Method qualification process can be related to equipment qualification step and further classified to installation qualification (IQ), operational qualification (OQ) and performance qualification (PQ).
Installation qualification of method may include few check points on instrument calibration status and qualification of analyst, training records. The detailed knowledge regarding method procedure, sample preparation, operating controls etc. should be conveyed to the chemist. In method operational qualification analysis ensures that all the operational parameters that have been optimized during method design phase operate as per requirement. This procedure is simply following ICH Q2 (R1) chapter. The method needs to be verified for parameters such as accuracy, precision, LOD, LOQ, linearity and range, ruggedness and robustness. All the results should be documented and analyzed. Method operational qualification is an additional assurance to support that the method operates as per defined ATP. Method performance qualification involves implementation of developed and validated method to actual routine analysis of commercial batches. The process demonstrates that the method consistently performs as per intended use with actual samples, facilities, instrument and analysis that will operate the method.
C. Stage 3-Control strategy design/Life cycle management
During the lifecycle of a product, both the manufacturing process and the method are likely to experience a number of changes brought through unintentional deviations, continuous improvement activities or the need to operate the method and\or process in a different environment. It is essential that all changes to the method operating conditions be considered in light of the knowledge and understanding that exists on the method performance. This activity aligns with the guidance in USP chapter 1010 on system performance verification. The goal of this stage is to assure that the optimized method consistently works as per the intended use. This phase includes eventual replication of optimized experiments, data collection and analysis to assure that the method remains in the state of control. An ongoing trend to collect and analyze data of method performance should be established to assure that method performs as per the intended use. Documenting this data will determine the degree of impact of planned or accidental changes on method. Close consideration should be given to all changes to the method operating region that results in any OOS or OOT. There should be a controlled strategy plan to identify the root cause. Corrective and preventive measures should also be defined.
If the method operating conditions are modified such that they fall outside the known method operable region then method should be redesigned. Likewise if the process changes and samples contain analytes at levels outside those covered in the method operable design region or have new interferences, a new ATP would need to be generated and a partial re-qualification exercise will be required to ensure the method is still capable of producing consistent and reliable results for the extended range or modified sample. Where a method needs to be transferred to new location, appropriate Method Installation Qualification activities (including knowledge transfer) will need to be performed in addition to a Method Qualification exercise.
Conclusion
An accurate data analysis tool is necessary to evaluate any process or system to assure that it works consistently as intended. Implementing QbD is one of the approaches that devoutly make scientist to understand the process or system closely. Optimizing process by QbD has become mandatory by some of the regulatory guidelines around the globe. Never the QbD approach can also be implemented successfully in analytical method development and optimization. Identifying critical parameters, performing risk assessment studies of critical parameters, using DOE for screening and method optimization are some of the milestones for QbD implementation, whenever magnificently applied will result in a robust method with fewer trials. Analyst also gains confidence in the method performance as the approach provides understanding between the method variables and performance. The overall advantage of the approach is improved method proficiency, reduced variability, less trials hence less method cost and reduced time consumption, knowledge about the extreme limitations of the method which when traversed may lead to method failures and at times method alternatives. Hence this approach can be well practiced for complexed chromatographic method where more number of analytes needs efficient separations.
References
- Peraman R, Bhadraya K and Reddy Y. Analytical Quality by Design: A tool for regulatory flexibility and robustness analytics. International journal of analytical chemistry. 2015: 1-9.
- Reid G L, Cheng G, Fortin D T, Harwood JW, Morgado JE, Wang J, et al. Reversed – phase liquid chromatographic method development in an analytical quality by design framework. Journal of liquid chromatography and related technologies. 2013: 36: 2612-2638.
- Bhatt D and Rane S. QbD approach to analytical RP-HPLC method development and its validation. International journal of pharmacy and pharmaceutical sciences. 2011: 3: 179-187.
- Karmarkar S, Garber R, Ghenchanok Y, George S, Yang X and Hammond R. Quality by design (QbD) based development of a stability indication HPLC method for drug and impurities. Journal of chromatographic science. 2011: 49: 439-446.
- International conference on Harmonization (ICH) Q8(R2): Pharmaceutical Development August 2009.
- International Conference on Harmonization (ICH) Q9: Quality Risk Management November 2005.
- International Conference on Harmonization (ICH) Q10: Pharmaceutical Quality System June 2008.
- Pharmaceutical CGMP’s for the 21th century – a risk based approach Final report, Department of Health and Human services USFDA, September 2004.
- Trivedi B. Quality by Design (QbD) in Pharmaceuticals. International Journal of Pharmacy and Pharmaceutical Sciences. 2012; 4: 17-29.