
Research Article
Austin J Anal Pharm Chem. 2017; 4(1): 1082.
High Performance Liquid Chromatographic Assay of Chlorpropamide, Stability Study and its Application to Pharmaceuticals and Urine Analysis
Basavaiah K¹* and Rajendraprasad N²
¹Department of Chemistry, University of Mysore, Mysuru, Karnataka, India
²PG Department of Chemistry, JSS College of Arts, Commerce & Science, B N Road, Mysuru, Karnataka, India
*Corresponding author: Basavaiah K, Department of Chemistry, University of Mysore, Manasagangothri, Mysuru, Karnataka, India
Received: February 27, 2017; Accepted: March 16, 2017; Published: March 22, 2017
Abstract
Chlorproamide (CLP) is a sulphonyl-urea derivative used in the treatment of type 2 diabetes mellitus. A rapid, sensitive and specific HPLC method with uv detection is described for the determination of CLP in bulk and tablet forms. The assay was performed on an Inertsil ODS 3V (150mm × 4.6mm; 5μm particle size) column using a mixture of phosphate buffer (pH 4.5), methanol and acetonitrile (30:63:7v/v/v) as mobile phase at a flow rate of 1mLmin-1 and with uv detection at 254nm. The column temperature was 30?C and injection volume was 20μL. The retention behaviour of CLP as a function of mobile phase pH, composition and flow rate was carefully studied, and chromatographic conditions, yielding a symmetric peak with highest number of theoretical plates, were optimized. The calibration curve was linear (r = 0.9999) over the concentration range 0.5 – 300 μgmL-1. The limits of detection (LOD) and quantification (LOQ) were found to be 0.1 and 0.3 μgmL-1, respectively. Both intra-day and inter-day precisions determined at three concentration levels were below 1.0% and the respective accuracies expressed as %RE were =1.10%. Assays were performed under slightly altered chromatographic conditions, and the results were not significantly different from those obtained under optimum conditions, reflecting the robustness of the method. Inter-equipment and interanalysts deviations were insignificant testifying the ruggedness of the method. The method was validated for selectivity via placebo blank and synthetic mixture analyses. The method was applied to determine CLP in tablets and the results were comparable with those obtained by a reference method with respect to accuracy and precision. As part of stress testing, CLP was subjected to forced degradation under acid and base-induced hydrolysis, oxidation, heat and light, followed by assay using the developed method, and the results indicated that the drug was prone to oxidative degradation, but inert to other stress-conditions. The method was sensitive and selective enough to determine the drug content in spiked urine, and the results were satisfactory.
Keywords: Chlorpropamide; Assay; HPLC; Pharmaceuticals; Stabilityindicating
Introduction
Chlorpropamide (CLP), chemically called 4-Chloro-N- [(propylamino) carbonyl]benzenesulfonamide (Figure 1), is one of the sulphonyl-urea derivatives, which are widely used as hyperglycemic drugs used in the treatment of non-insulin dependent diabetes mellitus [1]. CLP is found to stimulate pancreatic β-cell insulin production, which causes the reduction of glucose levels in blood.
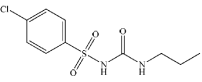
Figure 1: Structure of CLP.
Therapeutic use of the drug has resulted in the development of the many methods for its determination in body fluids and includes high performance liquid chromatography [2-7], gas-liquid chromatography [8-11], liquid chromatography-tandem mass spectrometry [12], micellar electrokinetic chromatography [13], electro spray mass spectrometry [14] and gas chromatography-mass spectrometry [15].
The drug has an official monograph in the United States Pharmacopeia [16] which has adopted an HPLC-uv detection method for the assay of CLP. British Pharmacopeia [17] describes a spectrophotometric method whereas European Pharmacopeia [18] offers a titrimetric method for the assay in tablets and bulk drug, respectively. In addition to the above, CLP in pharmaceuticals has been assayed by several other techniques such as titrimetry [19], spectrophotometry [20-23], electrophoresis [24], thinlayerchromatoghraphy [25, 26] and coulometry [27].
HPLC, which finds immense applications in the analysis of body fluids for CLP [2-7], has also been applied to the determination of drug in pharmaceuticals, chiefly due to its sensitivity, selectivity, speed and wide applicability. El-Gizawy [28] has reported an HPLC method for the determination of CLP in tablets. The author used a Zorbax SAX anion exchange column with phosphate buffer (pH 6.5) containing 1.7×10-2M β-cyclodextrin/methanol of ratio 85:15 as mobile phase. In a method described by Kumasaka et al. [29], a gradient RP-HPLC system equipped with a PAD using ODS column and acetonitrile-ammonium acetate buffer as a mobile phase, was employed to determine the drug in tablets with recovery in the range of 90.7- 105.7%. After homogenization of the sample with methanol, the drug in tablets was assayed by Molins et al. [30].
A quantitative HPLC method for the determination of CLP and its hydrolysis products: p-chlorobenzene sulphonamide and p-toluene sulphonamide, in solid dosage forms was developed by Robertson et al. [31]. However, the method was not applied for the assay of drug in tablets. Dasgupta [15] developed an RP-HPLC method for CLP in tablets. There was no interference from the products of hydrolysis and the excipients present in tablets.
Isocratic separation and simultaneous estimation of four antidiabetics belonging to sulphonyl urea family viz. CLP, to lbutamide, glipizide and gliclazide was achieved by Nadkarni et al. [32] using a Metaphase Bioscience column (250×4.6mm; 5μm) as stationary phase. The mobile phase was 0.02M KH2PO4 buffer, pH 3.0- methanol (40:60) with UV-detection at 230nm. A linear relationship was found in the range 0.4-10μg mL-1. The method was extended to the dosage forms of the four drugs.
The reported HPLC methods [28-32] have not been validated according to the current ICH guidelines and many of them suffer from low sensitivity and narrow linear ranges of applicability. None of the above method is stability-indicating, though stress testing is mandatory as per the ICH recommendations [33-35].
The objective of the present work was therefore, to develop a simple and sensitive HPLC method for the determination of CLP in pharmaceuticals, and to subject the drug to stress-testing via acid-, base-hydrolysis, oxidation, heating and photolysis to determine the stability-indicating ability of the developed method.
Experimental
Apparatus and software
Chromatographic analysis was performed with a Waters HPLC system (Waters Corporation, Milford, USA) equipped with Alliances 2695 series low pressure quaternary gradient pump, a programmable variable wavelength UV detector and auto sampler. Data were collected and processed using Waters Empower 2 software.
Materials and reagents
Pure CLP (99.9%) was kindly supplied by Deys Medical Stores (Mfg) Ltd, Kolkata, India, as a gift. Two brands of tablets containing CLP, Copamide-250 (Deys Medical Stores Ltd. Kolkata, India) and Diabinese-250 (Pfizer Pvt. Ltd. Mumbai, India) used in the investigation were purchased from local markets.
HPLC grade methanol and acetonitrile were purchased from Merck Ltd., India. Potassium dihydrogen orthophosphate, orthophosphoric acid, hydrochloric acid, sodium hydroxide and hydrogen peroxide were from Qualigens-India. Water purified by the Milli-Q system (Millipore, Milford, Massachusetts, USA) was used for mobile phase and sample diluent preparation. One litre of 10mM potassium dihydrogen orthophosphate solution was prepared by dissolving 1.4g of the salt in water and the pH was adjusted to 4.5 using triethylamine or dilute phosphoric acid. A 300mL portion of this buffer was mixed with 630mL of methanol and 70mL of acetonitrile, shaken well and filtered using 0.22μm Nylon membrane filter.
Hydrochloric acid (HCl, 0.1M) and hydrogen peroxide (H2O2, 5% v/v) were prepared by appropriate dilution of concentrated acid (Specific gravity 1.18) and commercial sample (30%) with water, respectively. Sodium hydroxide (NaOH, 0.1M) was prepared by dissolving the required quantity of chemical (S.D. Fine Chem Ltds, Bengaluru, India) in water.
Chromatographic conditions
Chromatographic assay was achieved on an Intersil ODS 3V (150mm ×4.6mm; 5μm particle size) column. A solution containing a mixture of phosphate buffer of pH 4.5, methanol and acetonitrile (30:63:7v/v/v) was used asa mobile phase. Mobile phase was used as a diluent. The flow rate was 1.0mLmin-1, the detector wavelength was set at 254nm and the injection volume was 20μL. The column temperature was maintained at 30°C.
Standard CLP solution
A stock standard solution containing 1000μg mL-1 CLP was prepared by dissolving accurately weighed 100mg of pure CLP in the mobile phase in a100mL calibrated flask.
General procedures
Procedure for bulk drug
Procedure for preparation of calibration curve: Working standard solutions containing 0.5-300μg mL-1 CLP were prepared by appropriate dilution of stock solution. Aliquots of 20μL were injected (triplicate) and eluted with the mobile phase under the stated chromatographic conditions. The average peak area versus concentration was plotted. Alternatively, the regression equation was derived using mean peak area-concentration data and concentration of unknown was computed from the regression equation.
Procedure for tablets: Tablet powder equivalent to 20mg CLP was transferred into a 100mLvolumetric flask containing 60mL of the mobile phase. The mixture was sonicated for 20min to achieve complete dissolution of CLP, and the content diluted to volume with the same solvent to yield a concentration of 200μg mL-1 CLP, and filtered through a 0.45μm nylon membrane filter. The tablet extract was injected on to the HPLC column in five replicates.
Procedure for placebo blank and synthetic mixture: A placebo blank of the composition of talc (15mg), starch (20mg), lactose (15mg), sodium alginate (20mg), calcium gluconate (15mg) and magnesium stearate (20mg) was made and its solution prepared as described under ‘assay procedure for tablets’ by taking about 20mg. To 10mg placebo, 10mg of pure CLP was added and homogenized, transferred to a 50mL calibrated flask and its extract prepared as described under “procedure for tablets”. Placebo blank and synthetic mixture solutions were injected on to the column in five replicates each.
Procedure for stress study: Ten mg of pure CLP was transferred separately into three different 50mLvolumetric flasks and dissolved with 10mL of mobile phase. Added 5mL of 0.1M HCl, 0.1M NaOH or 5% H2O2 separately, and the flasks were heated for 2h in a water bath maintained at 80°C. Then, the solutions were cooled, neutralized by adding base or acid and the volume in each flask was brought to the mark with mobile phase. An appropriate volume (20μL) was injected and chromatographed. Solid state thermal degradation was carried out by exposing pure drug to dry heat at 105°C for 2h. For photolytic degradation study, pure drug in solid state was exposed to 1.2 million lux hours in a photo stability chamber. The sample after exposure to heat and light was used to prepare 200μgmL-1 solutions in mobile phase separately, and the chromatographic procedure was followed.
Procedure for analysis of spiked human urine: Twenty mg of pure CLP was taken in a 50mL volumetric flask containing 10mL of drug free-urine, 5mL of mobile phase and 20mL of methanol were added and the contents were mixed well and the volume was brought to the mark with diluent. The solution was filtered through 0.22μm nylon membrane filter. Aliquots of 20μL were injected (n = 5) and eluted with the mobile phase under the stated chromatographic conditions after diluting to 100, 200 and 300μg mL-1 levels. The concentration of CLP was calculated using the regression equation, and percent recovery of CLP added determined.
Results and Discussion
With a view to achieve excellent linearity, sensitivity and selectivity, the method was optimized and validated in accordance with the current ICH guidelines [35]. The typical chromatograms obtained for blank and pure CLP under optimized HPL Cconditions are depicted in Figure 2.

Figure 2: Chromatograms for: a) Blank (mobile phase) b) Pure CLP solution
(200 μg mL-1).
Method development
The conditions affecting the retention behaviour of CLP were carefully studied and optimized so as to obtain satisfactory system suitability parameters such as retention time, number of theoretical plates and tailing factor. Mobile phase pH, composition and flow rate were the parameters of vital importance studied. The results of optimization of these parameters are compiled in Table 1.
Effect of pH and ionic strength of buffer
Mobile phases of pH ranging from 3.0-7.0, adjusted with sodium hydroxide or phosphoric acid were tried. The study revealed that the effect of pH of phosphate buffer was not critical over the pH range 4.5 to 6.5. Table 1 show that pH 4.5 was most appropriate and giving well defined peak and the highest number of theoretical plates. At lower and higher pH asymmetrical peak and smaller number of theoretical plates were observed.
The concentration of phosphate buffer
The effect of concentration of phosphate buffer on the chromatographic performance was investigated. Different concentrations in the range of 5 to 15mM were studied. The study revealed that the optimum chromatographic performance could be achieved with 10mM phosphate buffer. Although 5 and 8 mM gave the highest numbers of theoretical plates they were not selected due to the very poor reproducibility of retention times as well as the relatively high values of the tailing factors.
Type of organic modifier
Methanol or acetonitrile as organic modifier failed to yield a symmetric peak. A combination of methanol and acetonitrile (63:7 v/v) was the organic modifier of choice giving a symmetric and highly sensitive peak.
Ratio of organic modifier: The effect of ratio of organic modifier on the performance characteristics was investigated using mobile phases containing 30-63% methanol and 7-35% acetonitrile in different ratios. Table 1 shows that 63% methanol and 7% acetonitrile was the best, giving well defined peak and the highest number of theoretical plates. Different ratios of buffer and organic modifiers were tried and the optimum ratio was phosphate buffer, methanol and acetonitrile in the ration of 30:63:7v/v/v.
The effect of flow rate: The effect of flow rate on the symmetry, sensitivity and retention time of the peak was studied in the range of 0.5-1.5 mLmin-1 and a flow rate of 1.0 mLmin-1 was optimal for better symmetry and reasonable retention time (Table 1).
![]()
Ratio
(A/B/C)a
Number
of
theoretical
plates (N)
pH of
the
medium
Number
of
theoretical
plates (N)
Flow
rate,
mL
min-1
Number
of
theoretical
plates (N)
15/50/35
20/55/25
25/60/15
30/63/7
35/45/20
40/35/25
40/30/30
4212
5571
5685
6154
5718
5572
5015
3.0
3.5
4.5
5.5
6.5
6.8
7.0
3785
3893
6681
5954
5818
4236
4029
0.50
0.75
0.85
0.95
1.00
1.25
1.50
4172
4643
4795
5763
6199
5328
4927
aA: Phosphate buffer; B: Methanol and C-acetonitrile.
Table 1: Effect of ratio of organic modifier, pH and ionic strength of buffer on the number of theoretical plates.
Choice of column: Different columns including, Inertsil ODS 3V (150mm×4.6mm, 5μm particle size) column; Hypersil BDSC8 (250mm × 4.0mm, 5.0μm particle size) thermo column; Luna C18 (250mm×4.0mm, 5.0μm particle size); Chromatopack (250mm × 4.6mm, 5μm particle size) column and Zorbax XDB (250mm×4.0mm, 5.0μm particle size) column were tried for better performance characteristics. The first column (Inertsil ODS 3V) was found to be highly suitable with respect to suitability parameters.
Choice of detection wavelength: The UV detector response of CLP in methanol showed a maximum at 254nm with maximum absorptivity.
Method validation
Linearity: Linearity was studied by preparing standard solutions of different concentrations from 0.5 to 300 μg mL-1, plotting a graph of mean peak area against concentration and determining the linearity by least-square regression quation. The calibration plot was linear over the concentration range 0.5-300 μg mL-1 (n= 3) (Figure 3) and is described by the equation:
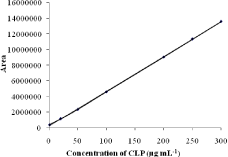
Figure 3: Calibration curve.
y = m x + b
where y is the mean peak area, x the concentration of CLP in μg mL-1, m slope and b intercept. The slope (m), y-intercept (b) and their standard deviations were calculated and are presented in Table 2. The calculated correlation coefficient (r = 0.9999) confirms the linear relationship between the peak areas and concentration of CLP.
![]()
Parameter
Value
Linear range, μg mL-1
Limits of detection, (LOD), μg mL-1
Limits of quantification, (LOQ), μg mL-1
Regression equation, y*
Slope (m)
Intercept (b)
Standard deviation of intercept (Sb)
Standard deviation of slope (Sm)
Correlation coefficient (r)
0.5 - 300
0.10
0.30
44395
211943
169.2
827.9
0.9999
*y=mx+b, where y is the mean peak area, x concentration in μg mL-1, b intercept, m slope.
Table 2: Linearity and regression parameters.
Precision and accuracy: Pure drug solution at three concentration levels was injected in seven replicates during the same day and over a period of five days (five injections per day) as part of intra-day and inter-day precision and accuracy study. Error and standard deviations were calculated for each data set. Pooled standard deviation was computed to determine the inter-day precision.
The percent relative error which is an indicator of accuracy is =1.1% and is indicative of high accuracy. The peak area-based and retention time-based %RSD values were <1% and reflective of very high repeatability and reproducibility of the developed method (Table 3).
![]()
CLP
injected,
μg mL-1
Intra-day (n = 7)
Inter-day (n = 5)
CLP
found,
μg mL-1
%
REa
%
RSDb
%
RSDc
CLP
found,
μg mL-1
%
REa
%
RSDb
%
RSDc
100
200
300
100.5
201.6
301.9
0.50
0.80
0.63
0.54
0.42
0.39
0.27
0.36
0.63
101.1
198.7
302.1
1.10
0.65
0.70
0.45
0.29
0.36
0.30
0.45
0.36
aRelative error; bRelative standard deviation based on peak area; cRelative standard deviation based on retention time.
Table 3: Results of accuracy and precision study.
Method robustness: Robustness of the method was evaluated by performing analysis under slightly altered chromatographic conditions like, detection wavelength, flow rate, mobile phase composition and column temperature, and the results were compared with those of the optimized chromatographic conditions. The result expressed as intermediate precision (%RSD) is summarized in Table 4 and speaks of the robustness of the method.
![]()
Condition
altered
Modification
Mean peak
area
± SD*
%
RSD
Mean Rt
± SD*
%
RSD
Mean
theoretical
plates
± SD*
%
RSD
Mean
tailing
factor
±SD*
%
RSD
Actual
-
9069466
± 43327
0.48
4.902±
0.004
0.082
6218±
6.439
0.10
1.109±
0.003
0.27
Column
temperature
30 and
30 ± 2 ?C
9067345
± 46618
0.54
4.921±
0.002
0.061
6155±
6.392
0.11
1.112±
0.004
0.36
Mobile
phase
composition
(Buffer:
methanol)
30:63:7
28:65:5
32:60:8
9067945
± 45414
0.51
4.909±
0.003
0.061
6236±
5.732
0.09
1.115±
0.003
0.27
Flow rate
1.0 and
1.0± 0.1 mL min-1
9068129
± 48196
0.53
4.918±
0.002
0.041
6189±
5.572
0.09
1.105±
0.004
0.36
Wavelength
254 and
254 nm ±
1 nm
9069992
± 52312
0.58
4.927±
0.004
0.081
6234±
6.963
0.11
1.101±
0.003
0.27
*Mean value of three determinations at CLP concentration of 200 μg mL-1.
Table 4: Results of method robustness reported as intermediate precision.
Method ruggedness: The ruggedness of the method was assessed by comparing results of assay performed by three analysts using the same column and also by a single analyst with three columns of the same type but belonging to different batches. The inter-analysts and inter-columns deviations were not significant. These results are presented in Table 5.
![]()
Variable
Mean
Peak
area
± SD*
%RSD
Mean
Rt ±
SD*
%RSD
Mean
theoretical
plates
±SD
%RSD
Mean
tailing
factor±
SD*
%RSD
Analysts
(n=3)
9067945
± 45414
0.51
4.918±
0.002
0.041
6234±
6.963
0.11
1.115±
0.003
0.27
Columns
(n = 3)
9278856
± 53628
0.58
5.216±
0.003
0.058
6196±
7.565
0.12
1.042±
0.004
0.38
*Mean value of three determinations at CLP concentration of 200 μg mL-1.
Table 5: Results of method ruggedness (n=3).
Selectivity: Selectivity of the method was evaluated by injecting the mobile phase, placebo blank, pure drug solution and tablet extract. No peaks were observed for mobile phase and placebo blank and no extra peaks were observed for tablet extracts (Figure 4). Synthetic mixture, when analysed at 200μgmL-1concentration level, yielded percent recoveries of 98.7% with standard deviation 0.81% indicating the absence of interference from the tablet excipients in the assay.
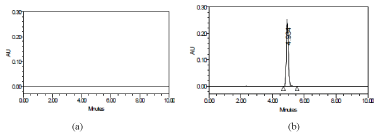
Figure 4: Chromatograms obtained for: a) placebo blank and b) tablet extract
(200μgmL-1 CLP).
Solution stability: The drug solution stored at lab temperature (25±2°C) was injected (n = 3) at different time intervals of 0, 12 and 24 h, and chromatograms were recorded. At the specified time interval, RSD (%) for the peak areas was calculated, and pooled standard deviation was computed for three sets of data generated at the end of 24h period. Low values of %RSD (=1%) are reflective of stability of solution over a period of 24h, and no significant change in the elution time of the peak and system suitability parameters: retention time, theoretical plates and tailing factor. The results of this study are compiled in Table 6.
![]()
Time, hour
Mean
Peak
area
± SD*
Pooled
%RSD
Mean
Rt ±
SD*
Pooled
%RSD
Mean
theoretical
plates
±SD
Pooled
%RSD
Mean
tailing
factor±
SD*
Pooled
%RSD
0
2784999
± 8319
0.62
5.575±
0.003
0.078
9019±
5.775
0.13
1.118±
0.003
0.41
12
2792397
± 9118
5.572±
0.004
9025±
6.192
1.122±
0.005
24
2812795
± 8217
5.583±
0.004
8954±
5.861
1.121±
0.004
*Mean value of three determinations for CLP concentration of 200 μg mL-1 at each time interval.
Table 6: Results of solution stability, reported as intermediate precision.
Application to tablets: The developed method was applied to the determination of CLP in two brands of tablets. The same tablet powder was assayed by European Pharmacopoeial method [18] for comparison. The comparison method is based on the titration of drug in alcohol against 0.1M sodium hydroxide to a phenolphthalein end point. The results were compared statistically by applying the Student’s test for accuracy and F-test for precision. As shown by the results compiled in Table 7, proposed method and the reference method do not differ significantly with respect to accuracy and precision, at the 95% confidence and for 4 degrees of freedom.
![]()
Formulation
brand
name
Nominal
amount,
mg
CLP found (%) ± SD (n = 5)
Reference
method
Proposed
method
t-value
F- value
Copamide
250
99.81±1.25
99.58±1.46
0.27
1.36
Diabenese
250
99.79±0.97
100.3±1.27
0.72
1.71
Tabulated t-value at 95% confidence level is 2.77; Tabulated F-value at 95% confidence level is 6.39.
Table 7: Results of determination of CLP in tablet and statistical comparison with the official method.
Accuracy by recovery test: To assess the accuracy and reliability of the method, recovery study via standard addition procedure was performed. To the pre-analyzed tablet powder, pure CLP was added at three levels and the total was determined by the proposed method. Each test was triplicated. When the test was performed on 250mg tablets, the percent recovery of pure CLP was in the range of 99.21– 101.7 with standard deviation values of 0.63-1.09. The results shown in Table 8 indicated that the method is very accurate and the common excipients found in tablet preparations did not interfere.
![]()
Tablet
studied
CLP in
tablet,
�g mL-1
Pure CLP
added,
�g mL-1
Total
found,
�g mL-1
Pure CLP
recovered*
(%NTG ±SD)
Copamide
99.58
50
150.9
102.6±1.04
99.58
100
202.9
103.3±0.81
99.58
150
247.6
98.68±1.09
Diabenese
100.3
50
152.6
104.6±1.05
100.3
100
201.3
101.0±0.63
100.3
150
248.9
99.07±0.72
*Mean value of three determinations.
Table 8: Results of recovery study by standard addition method.
Application to spiked urine: The developed and validated method was applied to determine CLP in spiked urine sample. The recovery of CLP was performed at three concentration levels. The results of this application compiled in Table 9 reveal that endogenous substances seldom interfere in the assay and that the method can be usefully applied to body fluids.
![]()
Spiked concentration
(μg mL-1)
Founda ±SD
%Recovery ±RSD
100
200
300
101.7±1.85
207.2±2.58
301.5±3.64
101.7±1.81
103.6±1.25
100.5±1.21
aMean value of five determinations; RSD is relative standard deviation.
Table 9: Results of CLP determination in spiked urine sample.
Results of forced degradation study
CLP was found to be stable under acidic, basic, thermal and photolytic stress conditions but slightly degraded under oxidative stress condition. The chromatograms obtained for CLP after subjecting to degradation are presented in Figure 5. The inference on degradation was based on comparison of the peak area of the stressed sample with the peak area of CLP sample without being subjected to stress study. The results of this study are shown in Table 10.
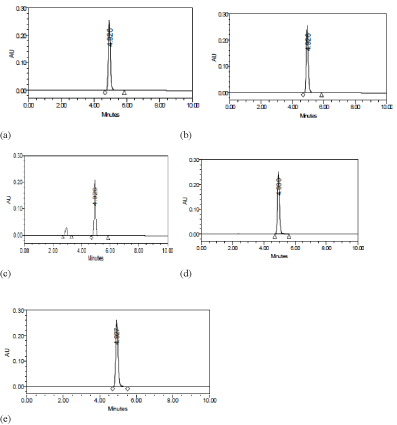
Figure 5: Chromatograms of CLP (200μgmL-1) after forced degradation a) acid degradation; b) base degradation; c) peroxide degradation; d) photolytic degradation
and e) thermal degradation.
![]()
Degradation condition
% Degradation
Acid hydrolysis
No degradation
Base hydrolysis
No degradation
Oxidation
19.2
Heat (105�C, 3 hours)
No degradation
Light (1.2 million lux hours)
No degradation
Table 10: Results of degradation study.
Conclusions
As mandated by the ICH guidelines, a stability indicating HPLC method was developed for the determination of chlorpropamide in pharmaceuticals and validated for linearity, LOD, LOQ, accuracy, precision, robustness, ruggedness and selectivity. This is the first report describing the stability-indicating assay of chlorpropamide. Compared to several methods reported earlier, the present method has a wide linear range enhancing its applicability to different levels of analyte in various matrices. A low detection limit of 0.1μgmL-1 makes the method one of the most sensitive ever developed for the drug in pharmaceuticals. Its application to spiked urine encourages the method to be applied to the determination of drug content in blood, plasma and serum after appropriate sample pre-treatment.
Acknowledgement
Authors thank Deys Medical Stores (Mfg) Ltd, Kolkata, India, for gifting chlorpropamide pure sample. Prof. K. Basavaiah is indebted to UGC, New Delhi, India, for financial assistance in the form of BSR Faculty fellowship.
References
- Diehl AK, Sugarek NJ and Bauer RL. Medication compliance in non-insulin-dependent diabetes: A randomized comparison of chlorpropamide and insulin. Diabetes Care. 1985; 8: 219-223.
- Naoya K, Farouk HS, Kaname O, Kimiko K, Mohamed MM, Kenichiro N, et al. HPLC determination of chlorpropamide in human serum by fluorogenicderivatization based on the suzuki coupling reaction with phenylboronic acid. Chromatographia. 2013; 76: 703-706.
- Bakare OMT, Enemali IS, Garba M and Obodozie OO. Rapid high performance liquid chromatographic determination of chlorpropamide in human plasma. African Journal of Biotechnology. 2007; 6: 1378-1381.
- Starkey BJ, Mould GP and Teale JD. The determination of sulphonylurea drugs by HPLC and its clinical application. Journal of Liquid Chromatography. 1989; 12: 1889-1896.
- Zecca L, Trivulzio S, Pinelli A, Colombo R and Tofanetti O. Determination of glibenclamide, chlorpropamide and tolbutamide in plasma by high performance liquid chromatography with ultraviolet detection. Journal of Chromatography B. Biomedical Sciences & Applications. 1985; 339: 203-209.
- Wåhlin-Boll E and Melander A. High-performance liquid chromatographic determination of glipizide and some other sulfonylurea drugs in serum. Journal of Chromatography. 1979; 164: 541-546.
- Hill RE and Crechiolo J. Determination of serum tolbutamide and chlorpropamide by high-performance liquid chromatography. Journal of Chromatography B. Biomedical Sciences & Applications. 1978; 145: 165-168.
- Midha KK, McGilveray IJ and Charette C. GLC determination of plasma levels of intact chlorpropamide or to lbutamide. Journal of Pharmaceutical Sciences. 1976; 65: 576-579.
- Chua HC, Stewart B, Lim BH and Lee HK. Screening of chlorpropamide in horse plasma by high-performance liquid chromatography with ultraviolet absorbance detection, and confirmation by gas chromatography–mass spectrometry. Journal of Chromatography B. Biomedical Sciences & Applications. 1998; 712: 243-252.
- Nasierowska Z, Suffczynski J, Szyszko E, Taton J, Kolinski P, Czech A, et al. Modification of the gas chromatographic method for blood chlorpropamide determination and evaluation of its use for clinical and pharmacological purposes. Polish Journal of Pharmacology & Pharmacy. 1983; 35: 405-415.
- Khalid S and Khawla S. Gas chromatographic method for determination of tolbutamide and chlorpropamide. Journal of Pharmaceutical Sciences. 1970; 59: 782-784.
- Guillaume H, Denis L, Thierry T, Arnaud R, Laurent B, Matthieu KL, et al. Identification and Quantification of 8 Sulfonylureas with Clinical Toxicology Interest by Liquid Chromatography–Ion-Trap Tandem Mass Spectrometry and Library Searching. Clinical Chemistry. 2005; 51: 1666-1672.
- Nunez M, Ferguson JE, Machacek D, Jacob G, Oda RP, Lawson GM, et al. Detection of hypoglycemic drugs in human urin using micellar electrokinetic chromatography. Analytical Chemistry. 1995; 67: 3668.
- Magni F, Marazzini L, Pereira S, Monti L and Kienle MG. Identification of sulfonylureas in serum by electrospray mass spectrometry. Analytical Biochemistry. 2000; 282: 136-141.
- Das Gupta. Quantitation of Chlorpropamide and Tolbutamide in Tablets by Stability-Indicating Reverse Phase High-Performance Liquid Chromatography. Analytical Letters. 1984; 17: 2119-2128.
- The United States Pharmacopoeia, 27th revision, US Pharmacopoeial Convention, MD, 2004, p 436.
- The British Pharmacopeia, Vol. III, Her Majesty’s Stationery Office, London, 2008, p 2530.
- The European Pharmacopeia 6.0. Vol. 01, 2008: 1087; p 1510.
- El-Bardicy MG, El-Khateeb SZ, Assaad HN and Ahmad AS. Mercurimetric determination of chlorpropamide by back titration. Indian Journal of Pharmaceutical Sciences. 1988; 50: 171.
- Lazaryan DS. IR spectrophotometric determination of cyclamide, chlorcyclamide, chlorpropamide and butamide. Farmatsiya. 1980; 29: 31-38.
- Mbah CJ and Okorie NH. Spectrophotometric Determination of Chlorpropamide in Bulk and Dosage Form by Complexation with Chloranilic Acid. Journal of Scientific Research. 2011; 3: 207-212.
- Zorya BP, Petrenko VV and Kulik IA. Spectrophotometric identification of some hypoglycemic drugs. Farmatsiya. 1989; 38: 69-71.
- Dnyanesh TN, Kasture AV. Separation and quantitative spectrophotometric estimation of phenformin hydrochloride and chlorpropamide in tablet dosage form. Indian Drugs. 2000; 37; 309-311.
- Ku YR, Chag LY, Ho LK and Lin JH. Analysis of synthetic anti-diabetic drugs in adulterated traditional Chinese medicines by high-performance capillary electrophoresis. Journal of Pharmaceutical and Biomedical Analysis. 2003; 33: 329-334.
- Nourrudin AW, Abdelwahab NS, El-Zeiny BA and Tohamy SI. Stability indicating TLC-densitometric method for determination of chlorpropamide. Journal of Liquid Chromatography and Related Technology. 2013; 36: 1575-1585.
- Anna G, Hanna H, Anna B and Dorota K. Normal- and reversed-phase thin-layer chromatography of seven oral antidiabetic agents. Journal of Planar Chromatography- Modern TLC. 2003; 16: 271-275.
- Kosta N, Slavka P, Milica B and Lucija A. Arhivza Farmaciju, 1992; 42: 53.
- El-Gizawy S. Bulletin of Faculty of Science. 1991; 20: 65.
- Kumasaka K, Kojima T, Honda H and Doi K. Screening and quantitative analysis for sulfonylurea-type oral anti-diabetic agents in adulterated health food using thin layer chromatography and high performance liquid chromatography. Journal of Health Sciences. 2005; 51: 453-460.
- Molins D, Wong CK, Cohen DM and Munnelly KP. High-pressure liquid chromatographic determination of chlorpropamide in tablet formulations. Journal of Pharmaceutical Sciences. 1975; 64: 123-124.
- Robertson DL, Butterfield AG, Kolasinski H, Lovering EG and Matsui FF. Stability-indicating high-performance liquid chromatographic determination of chlorpropamide, tolbutamide, and their respective sulfonamide degradates. Journal of Pharmaceutical Sciences. 1979; 68: 577-580.
- Nadkarni DR, Merchant RN, Sundaresan M and Bhagwat AM. Isocratic Separation and Simultaneous Estimation of Four Anti-Diabetic Members of the Sulphonyl Urea Family by Reverse Phase HPLC. Indian Drugs. 1997; 34: 650-653.
- ICH Harmonized tripartite guideline Q1A (R2), Stability testing of new drug substances and products, Geneva, February, 2003.
- International Conference on Hormonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Stability Testing: Photostability Testing of New Drug Substances and Products, Q1B, November, 1996.
- International Conference on Hormonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology Q2(R1), Complementary Guideline on Methodology dated 06 November 1996, incorporated in November 2005, London.