
Research Article
Austin J Anal Pharm Chem. 2017; 4(3): 1089.
Development of New Retinoids for Treatment of Epithelial Cancer
Mohanraj SM¹* and McMurry JE²
¹PolyMicrospheres, Division of Vasmo Inc., Indianapolis, IN, USA
²Baker Laboratory, Department of Chemistry, Cornell University, Ithaca, NY, USA
*Corresponding author: Mohanraj SM [1], PolyMicrospheres, Division of Vasmo Inc., 4101 East 30th Street, Indianapolis, IN 46218, USA
Received: July 29, 2017; Accepted: August 23, 2017; Published: August 30, 2017
Abstract
A new series of fifteen retinoids based on the dimethyltetralin ring system has been developed for evaluation as therapeutic agents in the treatment of epithelial cancer and other skin diseases. Four types of retinoids (A-D) were synthesized in which various side-chain double bonds are rigidly held in specific conformations, and their structure-activity relationships were investigated. Most of the new retinoids exhibit high activity in reversing keratinization in hamster tracheal organ culture. Retinoids 10b, 18a, and 18b show ED50(M) values in the (3-5)x10-11 range, and are promising candidates for the prevention and treatment of epithelial cancer. Among the most active retinoids, the 9E isomers are 40-60 times more active than the corresponding 9Z isomers.
Keywords: Aromatic retinoids; Cis-and trans-retinoids
Introduction
The retinoids, a large class of polyunsaturated diterpenes structurally related to vitamin A, have aroused much interest because of their diverse biological properties [2]. For example, vitamin A itself is important in promoting general growth, in regulating proliferation and differentiation of epithelial tissues, and in maintaining visual function and reproduction. The effect of vitamin A on epithelial tissues [3] has attracted much attention because vitamin A deficiency leads to hyperkeratosis of the skin and to metaplastic changes in the epithelia of gastrointestinal, respiratory, and urogenital tracts.
A number of synthetic retinoids have been reported to be extremely effective in the treatment of various types of keratinization disorders [3]. In addition, some synthetic retinoids exert antiinflammatory effects and seem to possess immunomodulatory properties [4], influencing dermal components such as lymphocytes and macrophages [2]. Retinoids have also been applied to the treatment of skin diseases like psoriasis and severe acne [3,5], and seem to interfere with the growth of oncogenic viruses and virus induced cancer [6].
One drawback to the use of known retinoids, both natural and synthetic, for the treatment of epithelial cancer and other keratinization disorders is that serious side effects often occur [7]. When given in high doses, retinoid levels rise in blood, tissues and liver, causing toxic effects known collectively as hypervitaminosis A syndrome. Though many retinoids have been synthesized, it still remains to the chemist to design and synthesize further structural variations of the retinoid skeleton that are more potent and less toxic, have improved pharmacokinetic properties, and are site specific.
The retinoic acid molecule is composed of three building units – a nonpolar cyclic end group, a polyene chain and a polar head group. Although it is relatively easy to probe structure-activity relationships involving the cyclic end group and polar head group, side-chain effects on biological activity are more difficult to study. The conformational flexibility of the side chain makes it possible for retinoids to adopt a large number of conformations, some of which are biologically active while others are inactive.
In order to probe the effects of side-chain conformation on biological activity, we decided to synthesize four types of new retinoids containing dimethyltetralin end groups (A-D in Chart 1), in which various double bonds are rigidly held in specific conformations. Few related structures with tetramethyltetralin end groups have been reported [8]. Our hope was to find a structure-activity relationship between side-chain conformation and activity that might allow chemists to design more suitable therapeutic agents. Bicyclic type A analogs have the 5,6 and 7,8 double bonds of the retinoic acid locked into an s-cis conformation without disturbing the rest of the molecule, while tricyclic analogs of types B–D have additional constraints on double bond geometry imposed by introduction of a third ring. In this paper, we report the synthesis and biological activity of these new retinoids and their 9Z isomers.
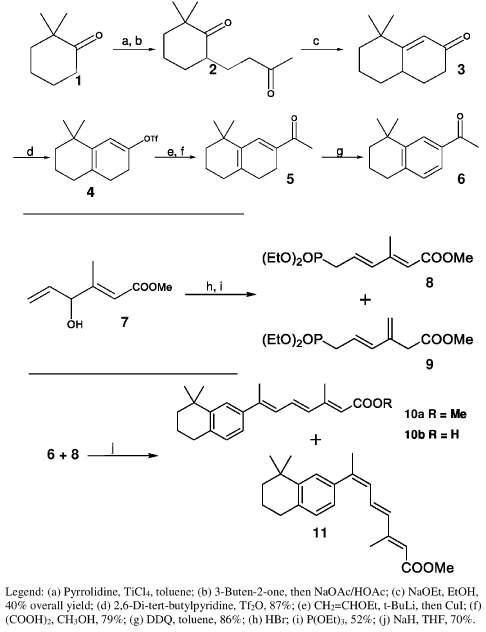
Chart 1: Structures of new Retinoids.
(Type A: 10-11, Type B: 15-16, Type C: 18-19, and Type D: 21-22).
Results and Discussion
Synthesis
Our plan was to prepare 2-acetyl-8,8-dimethyltetralin (6), the key intermediate necessary for the synthesis of the new retinoids, by Robinson annulation of 2,2-dimethylcyclohexanone, followed by introduction of the acetyl side chain and aromatization. Unfortunately, standard annulation methods using 3-buten-2-one under various basic or acidic conditions did not yield the desired bicyclic enone 3. Similarly, reaction of the trimethylsilyl ether derivative of 2,2-dimethylcyclohexanone with 3-buten-2-one in presence of either TiCl4 or TiCl4 / Ti(O-isoPr)4 yielded none of the expected diketone 2.
Our successful synthesis of tetralin 6 is shown in Scheme 1. Reaction of 2,2-dimethylcyclohexanone with pyrrolidine in the presence of TiCl4 gave an enamine, which, on treatment with 3-buten- 2-one, gave diketone 2. Cyclization of the diketone with NaOEt gave the required bicyclic enone 3. Enone 3 was next converted into 2-acetyl-3,4-dihydro-8,8-dimethyltetralin (5) through a sequence of steps beginning with formation of enol triflate 4. Treatment of ketone 3 with 2,6-di-tert-butylpyridine and triflic anhydride [9] gave 4, and coupling of 4 with ethoxyvinylcuprate [10] according to our previously published method [11], followed by enol ether hydrolysis, gave dienone 5. Oxidation with DDQ then provided the aromatic ketone 6.
Scheme 1: Synthesis of key intermediate 6 and retinoids 10-11 (Type A).
With ketone 6 thus available to act as a precursor of all target retinoids, we next synthesized the phosphonate intermediate 8 needed for the synthesis of type A retinoids. Methyl (E)-4-hydroxy- 2,5-hexadienoate (7) was prepared according to the procedure of Davalian and Heathcock [12]. Treatment of 7 with HBr followed by P(OEt)3 gave a mixture of phosphonates 8 and 9, which were separated quantitatively by HPLC using the peak shaving-recycling technique [13]. Various conditions were tried to optimize the Horner- Emmons reaction of phosphonate 8 with the key intermediate 6, but treatment of phosphonate 8 with NaH at -10°C for 7 minutes to form the sensitive anion, followed by reaction with acetyl tetralin 6 gave the best yield of the desired retinoids 10a and 11(type A). After separation of the ester mixture by HPLC [14], saponification of 10a gave acid 10b.
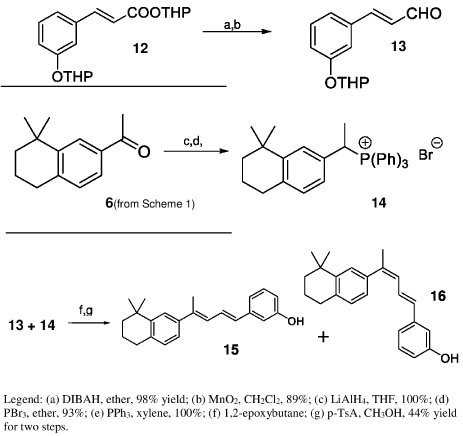
Scheme 2 shows the synthesis of type B retinoids. Selective reduction of tetrahydropyran-protected ether ester 12 with diisobutylaluminum hydride at 0°C followed by oxidation of the alcohol with activated MnO2, gave ether aldehyde 13. This aldehyde was then allowed to react with phosphonium salt 14, prepared from ketone 6 by LiAlH4 reduction, conversion into corresponding bromide and displacement with triphenylphosphine. The Wittig reaction was best carried out in the presence of 1,2-epoxybutane, which acts both as a mild base and as solvent, to yield a mixture of tetrahydropyran protected retinoids. Cleavage of the ether group by reaction with methanolic p-toluenesulfonic acid then gave retinoids 15 and 16 (type B), which were separated by HPLC [14].
Scheme 2: Synthesis of retinoids 15-16 (Type B).
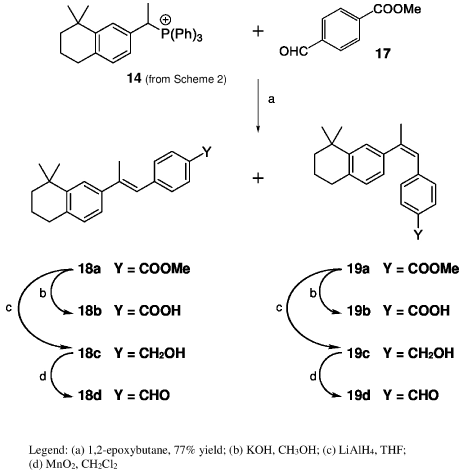
Type C retinoids were synthesized by the route in Scheme 3. Wittig reaction of phosphonium salt 14 with methyl p-formylbenzoate (17) was best carried out using 1,2-epoxybutane to obtain a 87:13 mixture of 18a and 19a. Irradiation of this mixture gave a 2:3 mixture of E and Z isomers, which were separated by HPLC [14] to give the pure esters. Saponification of these esters 18a and 19a gave the corresponding acids 18b and 19b, and LiAlH4 reduction gave the alcohols 18c and 19c. Oxidation with activated MnO2 then provided retinoid aldehydes 18d and 19d.
Scheme 3: Synthesis of retinoids 18a-19d (Type C).
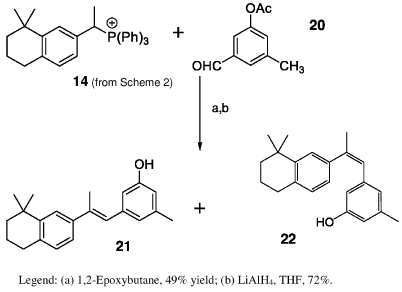
Scheme 4 shows the synthesis of type D retinoids. Wittig reaction of phosphonium salt 14 with commercially available aldehyde 20 was carried out using 1,2-epoxybutane to give a mixture of isomeric retinoid acetates, which, on treatment with LiAlH4 followed by HPLC separation [14], gave 21 and 22.
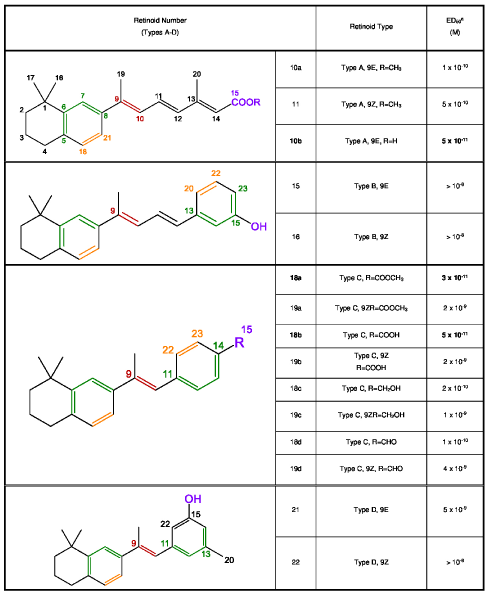
Scheme 4: Synthesis of retinoids 21-22 (Type D).
Nomenclature: Retinoid nomenclature follows IUPAC rules, using 5,6,7,8-tetrahydronaphthalene as the base ring system. Numbering of skeletal atoms is based on standard retinoid numbering. The naming and numbering the new synthetic retinoids was done with assistance from the editor of Nomenclature, Chemical Abstract Service [16].
Biological activity
Biological activity of all new synthetic retinoids was evaluated using the hamster tracheal organ culture (TOC) assay reported by Newton, Henderson and Sporn [15]. Since the TOC assay measures the intrinsic ability of retinoids to control epithelial cell differentiation, it is believed to have significant predictive value for the potential use of a new retinoid for the prevention and treatment of epithelial cancer.
Table 1 shows ED50(M) values of the new retinoids in the hamster tracheal organ culture assay (see Experimental Section). ED50(M) is the dose for reversal of keratinization in epithelium of 50% retinoid deficient hamster tracheas in organ culture. Dose-response curves were made for the reference substance “all-trans-retinoic acid” and each new retinoid, and from these data 50% effective dose ED50(M) was determined.
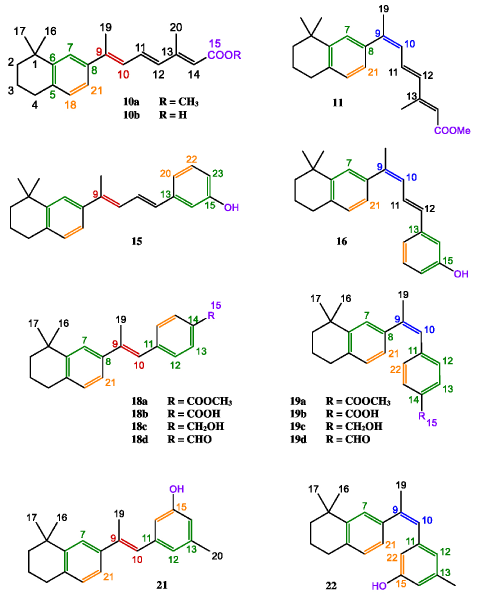
Table 1: Activity of new Retinoids in the TOC Assay.
Type A and C retinoids are more active than type B and D. The three most promising agents in this new series are retinoids 18a [ED50(M) = 3x10-11], 18b [ED50(M) = 5x10-11] and 10b [ED50(M) = 5x10-11]. Among type A retinoids, acid 10b is two times more active than the corresponding ester 10a. Among type C retinoids, the ratio of activity of ester (18a) / acid (18b) / aldehyde (18d) / alcohol (18c) = 6: 4: 2: 1.
In all cases, 9-trans retinoids (E isomers) are more active than their 9-cis retinoids (Z isomers). Comparing the ED50(M) values of the E and Z isomers of the most active retinoids, the 9E isomer 18a is 60 times more active than the 9Z isomer 19a (Type C, R=COOCH3) and the 9E isomer 18b is 40 times more active than the 9Z isomer 19b (Type C, R=COOH). In the case of the less active retinoids, the 9E isomer 10a is 5 times more active than the 9Z isomer 11 (Type A, R=CH3), the E isomer 18c is five times more active than the 9Z isomer 19c (Type C, R=CH2OH), and the E isomer 18d is 3.3 times more active than the 9Z isomer 19d (Type C, R=CHO).
Experimental Section
General
IR spectra were recorded on neat samples with a Perkin-Elmer 298 spectrophotometer, and UV spectra were taken in EtOH using a Perkin-Elmer 552A UV/VIS instrument. 1H NMR spectra were run at 300MHz on a Bruker WM 300 spectrophotometer in CDCl3 with Me4Si as internal standard. 13C NMR spectra were run at 22.49 MHz on a JEOL FX 90Q spectrometer in CDCl3 with Me4Si as internal standard. Chemical shifts were reported in ppm relative to Me4Si (d=0). High Resolution Mass Spectra (HRMS) were obtained on an AEI-MS-902 instrument at 70eV. HPLC separations were carried out on a Waters ALC 201 liquid chromatograph using a Whatman Partisil M9 10/50 column or on a Waters Prep LC / System 500A liquid chromatograph using two 5.7 x 30 cm Prep Pak 500 / silica cartridges. GC separations were carried out on a Gow Mac gas chromatograph with series 550 thermal conductivity detector using a 6’ x ¼” column packed with 10% SP2100 on 80/100 Supelcoport. All reactions were carried out under an atmosphere of argon. The phrase “worked up in the usual manner” refers to partitioning the crude reaction mixture between water and organic solvent, washing the organic layer with brine, drying over MgSO4, and concentrating on the rotary evaporator.
The 9E and 9Z isomers of retinoids (Type A-D, Table 1) were separated by HPLC [14]. The new retinoids were fully characterized using IR spectra, high resolution 1H and 13C NMR spectra, UV spectra, and High Resolution Mass Spectra (HRMS). In addition, a complete assignment of the 1H and 13C NMR signals of the retinoids to specific hydrogen and carbon atoms was achieved.
Dimethylhexahydronaphthalenone 3
2,2-Dimethylcyclohexanone (12.6g, 0.1mol) and pyrrolidine (50.1mL) were dissolved in 450mL toluene, and a solution of TiCl4 (7.7mL, 0.07mol) in 80mL toluene was added dropwise to the reaction mixture at -15°C over a period of 1h. The reaction mixture was allowed to warm to room temperature and stirred at 80°C for 12h, with monitoring by GC. The reaction mixture was then dried with anhydrous MgSO4 and concentrated to yield the enamine: bp 76-78°C (0.6mm); IR (neat) 1621, 1450, 1160cm-1; NMR d 4.55 (t, 1H, J = 4 Hz), 2.87 (tbr, 4H, J = 4.5 Hz), 2.08 (m, 2H), 1.77 and 1.52 (m, 8H) and 1.17 (s, 6H).
The enamine from the above reaction was dissolved in 80 mL tetrahydrofuran, filtered under argon, and cooled to -78°C. 3-Buten- 2-one (9.7mL, 0.12mol) was added dropwise, and the reaction mixture was brought to reflux. After 12h, a buffer solution of 4g NaOAc, 8mL water and 8mL HOAc was added, and the mixture was further refluxed for 4h. The reaction mixture was then diluted with water and extracted with diethyl ether. The ether extracts were combined, washed with 5% HCL, saturated NaHCO3 and brine, and concentrated to yield diketone 2 (ca 8g): IR 1705 (br) cm-1; NMR d 2.48 (m, 3H), 2.13 (s, 3H), 1.88 (m, 2H), 1.68 (m, 6H), 1.18 (s, 3H), and 1.05 (s, 3H).
Diketone 2 in 60 mL EtOH was added to a solution of sodium ethoxide (3.0 g) in 60mL EtOH and stirred at 60°C for 2h. After cooling and dilution with cold water, the reaction mixture was extracted with diethyl ether, concentrated, and purified by chromatography on silica gel to provide 6.92g (40% overall yield from 2,2-dimethylcyclohexanone) of bicyclic enone 3 and 0.364 g (2% overall yield) of the corresponding non-conjugated enone isomer. Data for enone 3: mp 71-73°C; IR 1672, 1610, 1455 cm-1; 1H NMR 5.94 (d, 1H, J = 1.8), 2.53 (m, 1H), 2.32 (m, 2H), 2.1 (m, 1H), 1.94 (m, 1H), 1.59 (m, 6H), and 1.14 (s, 6H); 13C NMR 199.94 (s, C2 carbonyl), 173.42 (s, C8a), 121.34 (d, C1), 40.29 (t, C7), 36.90 (s, C8), 35.76 (t, C3), 34.51 (t, C4), 34.16 (d, C4a), 29.03 (t, C5), 28.20 and 27.42 (q, C9 and C10 methyls), and 20.92 (t, C6); m/z (%) , 178 (M+, C12H18O, 100), 163 (22), 150 (33), 135 (36), 122 (50), 109 (70).
2-Acetyl-3,4,5,6,7,8-hexahydro-8,8-dimethylnaphthalene (5)
2,6-Di-tert-butylpyridine (0.9mL, 4.0mmol) and triflic anhydride (Tf2O, 0.8mL, 4.5mmol) were added to a solution of enone 3 (0.523g, 2.94mmol) in 15mL CH2Cl2 at 0°C, and the solution was stirred at room temperature for 16h. Solvent was then removed and residue was taken up in pentane, filtered, and washed with pentane. Concentration of the pentane solution and purification of the residue by chromatography on silica gel gave 0.788g (87% yield) of the triflate 4: IR 1670, 1610, 1428, 1220, 1140 cm-1; NMR 5.93 (s br, 1H), 2.42 (t br, 4H, J = 6), 1.98 (m, 2H), 1.55 (m, 4H), and 1.00 (s, 6H).
tert-BuLi (12.43mL of 2.1M solution in pentane, 26mmol) was added dropwise to a solution of ethyl vinyl ether (1.88g, 26mmol) in 17mL tetrahydrofuran at -78°C, and the resulting mixture was warmed to 5°C and stirred for 1.5h. The reaction mixture was cooled to -78°C and quickly transferred by cannula to a stirred suspension of CuI (1.657g, 9mmol) in 43mL tetrahydrofuran at -78°C. The reaction mixture slowly became deep red as it was warmed to -25°C and held there for 2h. A solution of triflate 4 (0.54g, 2mmol) in 3mL tetrahydrofuran was added dropwise at -25°C, and the reaction mixture was warmed to room temperature and stirred for 12h. The reaction mixture was then filtered through beds of silica gel and Florisil, washed with diethyl ether, and concentrated. The residue was stirred with 100mL of 0.1M oxalic acid in methanol for 1h and purified by chromatography on silica gel to furnish 0.282g (79% yield from triflate 4) of 2-acetyl-3,4,5,6,7,8-hexahydro-8,8-dimethylnaphthalene (5): mp 28-29°C; IR 1650, 1570, 1250 cm-1; 1H NMR 7.02 (s br, H-1), 2.36 (t br, J = 8.8, H-3), 2.33 (s, acetyl methyl), 2.09 (t, J = 6.6, H-5), 2.08 (t, J = 8.2, H-4), 1.64 (m, H-6), 1.52 (dt, J = 2.2 and 5.5, H-7), and 1.07 (s, C9 and C10 methyls); 13C NMR 198.11 (s, acetyl carbonyl), 139.31 (s, C8a), 135.36 (d, C1), 134.63 (s, C4a), 134.44 (s, C2), 38.78 (t, C7), 32.44 (s, C8), 31.41 (t, C4), 29.22 (t, C5), 28.49 (q, C9 and C10 methyls), 24.98 (q, acetyl methyl), 20.10 (t, C3), and 19.03 (t, C6); m/z (%), 204 (M+, C14H20O, 43), 189 (39), 147 (27), 91 (26), 43 (100).
2-Acetyl-5,6,7,8-tetrahydro-8,8-dimethylnaphthalene (6)
A mixture of dienone 5 (1.802g, 8.83mmol) and dichlorodicyanobenzoquinone (DDQ, 2.2g, 9.7mmol) in 110mL toluene was stirred at 82°C for 18h. The resulting ketone was purified by chromatography on silica gel to afford 1.523g (86% yield) of 2-acetyl-5,6,7,8-tetrahydro-8,8-dimethylnaphthalene (6): IR 1672, 1596, 1350, 1245 cm-1; 1H NMR 7.95 (d, J = 1.8, H-1), 7.63 (dd, J = 1.8 and 8, H-3), 7.11 (d, J = 7.9, H-4), 2.81 (t, J = 6.3, H-5), 2.57 (s, acetyl methyl), 1.81 (m, H-6), 1.68 (dt, J = 2.3 and 5.9, H-7), and 1.31 (s, C9 and C10 methyls); 13C NMR 181.83 (s, acetyl carbonyl), 146.28 (s, C8a), 142.19 (s, C4a), 135.22 (s, C2), 129.27 and 126.68 (d, C4 and C1), 125.32 (d, C3), 39.02 (t, C7), 34.00 (s, C8), 31.71 (q, C9 and C10 methyls), 30.93 (t, C5), 26.49 (q, acetyl methyl), and 19.32 (t, C6); m/z (%), 202 (M+, C14H18O, 23), 187 (100), 43(88). HRMS calc’d for C14H18O 202.1363, found 202.1368.
Methyl (2E, 4E, 6E)- and methyl (2E, 4E, 6Z)-3-methyl- 7-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)-2,4,6- octatrienoates (10a and 11)
A solution of 0.872g methyl (E)-4-hydroxy-3-methyl-2,5- hexadienoate (7) in 10mL diethyl ether was prepared by the method of Davalian and Heathcock [14] and added dropwise at -20°C to a saturated solution of anhydrous HBr in 20mL diethyl ether. The reaction mixture was warmed to -10°C and stirred for 2h, then warmed to room temperature and stirred for 30min. Workup in the usual manner gave 1.13g of an oily residue that was taken up in 1.5mL of P(OEt)3 and stirred at 140°C for 1.75h. Chromatography of the reaction mixture on silica gel gave a mixture of phosphonates, which were separated by HPLC to afford 0.525 g (34% yield) of 8 and 0.278 g (18% yield) of 9. Data for methyl (2E, 4E)-6-(diethylphosphono)- 3-methyl-2,4-hexadienoate (8): IR 1710, 1610, 1240, 1158, 1050, 1025, 960 cm-1; NMR 6.24 (dd, J = 4.9 and 15.7, H-4), 6.06 (m, H-5), 5.75 (s br, H-2), 4.11 (q, J = 7.5, phosphonate methylenes), 3.71 (s, -COOCH3), 2.72 (dd, J = 7.4 and 22.7, H-6), 2.28 (s br, C3 methyl), 1.32 (t, J = 7.2, phosphonate methyls); m/z (%), 276 (M+, C12H21O5P, 16), 138 (100), 125 (29), 79 (37). Data for 9: IR 1740, 1605 (w), 1250, 1155, 1020, 960 cm-1; NMR 6.25 (dd, J = 4.9 and 15.8, H-4), 5.64 (m, H-5), 5.18 and 5.11 (s br, C3 methylene), 4.10 (q, J = 7.4, phosphonate methylenes), 3.69 (s, -COOCH3), 3.24 (s, H-2), 2.65 (dd, J = 7.3 and 22, H-6), and 1.32 (t, J = 7, phosphonate methyls); m/z (%), 276 (M+, C12H21O5P, 40), 138 (59), 79 (100).
Phosphonate 8 (169mg, 0.612mmol) in 2mL tetrahydrofuran (THF) was added to a slurry of NaH (36mg of 50% dispersion in oil, 0.75mmol) in 1mL THF and the mixture was stirred at -10°C for 7min. A solution of enone 6 (69mg, 0.34mmol) in 2mL THF was then added and the reaction was further stirred at -10 to 0°C for 3h and at room temperature for 16h. The reaction mixture was diluted with water and extracted with diethyl ether. The ether extract was washed with water and brine, dried and evaporated. Purification of the residue by chromatography on silica gel gave 77mg (70% yield) of E and Z retinoid esters 10a and 11 (65:35), which were separated by HPLC. Data for methyl (2E, 4E, 6E)-3-methyl-7-(5,6,7,8-tetrahydro- 8,8-dimethyl-2-naphthyl)-2,4,6-octatrienoate (10a): IR 1710, 1602, 1590, 1150 cm-1; 1H NMR 7.42 (d, J = 1.8, 1H), 7.19 (dd, J = 1.9 and 8.0, 1H), 7.04 (dd, J = 11.3 and 15.1, 1H) 7.03 (d, J = 8.1, 1H), 6.54 (d br, J = 11.1, 1H), 6.38 (d, J = 15.2, 1H), 5.81 (s br, 1H), 3.72 (s, 3H), 2.77 (t, J = 6.2, 2H), 2.39 (d, J = 0.8, 3H), 2.25 (d, J = 0.8, 3H), 1.80 (m, 2H), 1.67 (m, 2H), and 1.31 (s, 6H); 13C NMR 167.49 (s), 152.96 (s), 145.70 (s), 140.68 (s), 140.09 (s), 136.04 (s), 135.41 (d), 131.36 (d), 129.07 (d), 125.76 (d), 123.90 (d), 122.83 (d), 118.15 (d), 50.92 (q), 39.31 (t), 33.95 (s), 31.85 (q, 2C), 30.49 (t), 19.61 (t), 16.44 (q), and 13.86 (q); UV 347 (42020), 254 (11020), 210 (s), 202 nm (23650); HRMS calc’d for C22H28O2 324.2089, found 324.2104. Data for methyl (2E, 4E, 6Z)-3-methyl-7-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)-2,4,6- octatrienoate (11): IR 1710, 1600, 1590, 1150 cm-1; 1H NMR 7.20 (d, J = 1.6, 1H), 7.06 (d, J = 8.1, 1H), 6.98 (dd, J = 1.7 and 7.9, 1H), 6.80 (dd, J = 11.1 and 15.2, 1H), 6.26 (d, J = 15.1, 1H), 6.23 (d, J = 11.0, 1H), 5.75 (s br, 1H), 3.69 (s, 3H), 2.78 (t, J = 6.0, 2H), 2.18 (s, 6H), 1.81 (m, 2H), 1.68 (dt, J = 2.2 and 5.7, 2H), and 1.29 (s, 6H); 13C NMR 167.54 (s), 153.30 (s), 145.41 (s), 143.55 (s), 138.19 (s), 135.70 (s), 134.19 (d), 132.97 (d), 128.98 (d), 127.12 (d), 126.59 (d), 124.73 (d), 117.71 (d), 50.87 (q), 39.22 (t), 33.85 (s), 31.80 (q, 2C), 30.54 (t), 25.51 (q), 19.61 (t), and 13.77 (q); UV 333.5 (36010), 255.5 (12400), 208 (s), 202 nm (28790); HRMS calc’d for C22H28O2 324.2089, found 324.2098.
Hydrolysis of ester 10a (22 mg) was accomplished by treatment with 10mL of 2N KOH in 2mL water and 8mL methanol at 50°C for 3h. Acidification of the reaction mixture at 0°C with HCl and extraction with chloroform, and recrystallization from ethyl acetatehexane gave 19mg (90% yield) of (2E, 4E, 6E)-3-methyl-7-(5,6,7,8- tetrahydro-8,8-dimethyl-2-naphthyl)-2,4,6-octatrienoic acid (10b): mp 202-203 °C; IR 3200-2300, 1680, 1600, 1580, 1220, 1180 cm-1; 1H NMR 7.43 (d, J = 1.7, 1H), 7.20 (dd, J = 1.8 and 7.9, 1H), 7.08 (dd, J = 11.1 and 14.9, 1H), 7.04 (d, J = 8.0, 1H), 6.56 (d br, J = 11.2, 1H), 6.41 (d, J = 15.1, 1H), 5.84 (s br, 1H), 2.77 (t, J = 6.2, 2H), 2.40 (s, 3H), 2.26 (s, 3H), 1.80 (m, 2H), 1.67 (dt, J = 2 and 5.8, 2H), and 1.32 (s, 6H); 13C NMR 172.13 (s), 155.30 (s), 145.80 (s), 141.36 (s), 140.04 (s), 136.24 (s), 135.26 (d), 132.14 (d), 129.12 (d), 125.76 (d), 124.00 (d), 122.88 (d), 117.71 (d), 39.31 (t), 34.00 (s), 30.54 (t), 31.90 (q, 2C), 19.66 (t), 16.54 (q), and 14.11 (q); UV 337 (47980), 250 (13140), 201.5 nm (31600); HRMS calc’d for C21H26O2 310.1933, found 310.1950.
Tetrahydropyranyl m-tetrahydropyranyloxycinnamate (12)
Dihydropyran (DHP, 5.6mL) was added to a solution of m-hydroxycinnamic acid (1g, 6.7mmol) in 10mL diethyl ether. The mixture was cooled to 0°C, 19mg of p-toluenesulfonic acid was added, and the solution was stirred at room temperature for 16h. Workup in the usual manner with diethyl ether gave 2.15g (97% yield) of tetrahydropyran (THP) protected ether ester 12: IR 1715, 1635, 1580, 1445, 1360-860 cm-1; NMR 7.70 (d, J = 15.8, vinyl H next to aromatic ring), 7.26 (m, 2H), 7.15 (d, 1H, J = 7.7), 7.08 (m, 1H), 6.45 (d, J = 15.8, vinyl H next to carboxyl), 6.10 (t, J = 3, H-2 of ester THP), 5.44 (t, J = 3, H-2 of ether THP), 3.92 (m, H-6 of ester THP), 3.72 and 3.61 (m, H-6 of ether THP), 1.97, 1.86 and 1.66 (m, 12H).
m-Tetrahydropyranyloxycinnamaldehyde (13)
Selective reduction of the ester group of compound 12 (0.258g) in diethyl ether with di-isobutylaluminum hydride (DIBAH) at 0°C for 30min, followed by workup in the usual manner gave 0.178g (98% yield) of the corresponding alcohol: IR 3400, 1600, 1580, 1290-960 cm-1; NMR 7.07 (m, 4H), 6.70 (d, J = 21.5, vinyl H next to aromatic ring), 6.43 (dt, J = 4.5 and 21.5, vinyl H next to CH2OH), 5.42 (t br, H-2 of ether THP), 4.27 (d, J = 4.5, -CH2OH), 3.77 (m, H-6 ether THP), 2.75 (br, -CH2OH), and 1.77 (m, 6H).
Reaction of this alcohol (158mg) in 30mL dichloromethane with 790mg of activated MnO2 at room temperature for 12h, followed by passage through a bed of activated charcoal and Florisil gave 139mg (89% yield) of aldehyde 13: IR 1682, 1630, 1580, 1270, 1125 cm- 1; NMR 9.67 (d, J = 7.7, -CHO), 7.42 (d, J = 15.8, vinyl H next to aromatic ring), 7.29 (m, 2H), 7.15 (m, 2H), 6.68 (dd, J = 7.7 and 15.8, vinyl H next to CHO), 5.44 (t, J = 3.3, H-2 of ether THP), 3.88 and 3.61(m, H-6 of ether THP), 1.99 (m, 1H), 1.87 (m, 2H), and 1.66 (m, 3H).
[1-(5,6,7,8-Tetrahydro-8,8-dimethyl-2-naphthyl)-ethyl]- triphenylphosphonium bromide (14)
The key intermediate 6 (189mg) was reduced with LiAlH4 in usual manner to afford 191mg (100% yield) of the corresponding alcohol: IR 3360,1620 (w), 1580 (w), 1460, 1370, 1090 cm-1; 1H NMR 7.31 (d, J = 1.5, H-1), 7.06 (dd, J = 1.8 and 7.7, H-3), 7.00 (d, J = 7.7, H-4), 4.80 (q, J = 6.6, -CHOH-CH3), 2.73 (t, J = 6.3, H-5), 1.78 (m, H-6), 1.65 (m, H-7), 1.46 (d, J = 6.6, -CHOH-CH3), and 1.28 (s, C9 and C10 methyls); 13C NMR 145.59 (s, C8a), 143.44 (s, C4a), 135.01 (s, C2), 129.06 and 123.55 (d, C4 and C1), 122.43 (d, C3), 70.21 (d, -CHOHCH 3), 39.40 (t, C7), 33.89 (s, C8), 31.84 (q, C9 and C10 methyls), 30.47 (t, C5), 25.10 (q, -CHOH-CH3), and 19.70 (t, C6); m/z (%), 204 (M+, C14H20O, 25), 189 (100), 145 (37), 91 (31).
PBr3 (0.59mL, 6.29mmol) was added dropwise to a solution of the above alcohol (0.642g, 3.15mmol) in 15mL ether at 0°C, and the mixture was stirred for 1h. The usual workup with diethyl ether gave 0.776g (93% yield) of bromide: IR 1610 (w), 1570 (w), 1500, 1455, 1360, 1215, 1178, 818, 650 cm-1; 1H NMR 7.35 (d, J = 1.8, H-1), 7.15 (dd, J = 1.8 and 7.7, H-3), 7.00 (d, J = 7.7, H-4), 5.18 (q, J = 7, -CHBr- CH3), 2.72 (t, J = 6.3, H-5), 2.02 (d, J = 7, -CHBr-CH3), 1.77 (m, H-6), 1.64 ( m, H-7), 1.27 and 1.28 (s, C9 and C10 methyls); 13C NMR 145.84 (s, C8a), 140.58 (s, C4a), 136.29 (s, C2), 129.27 and 124.93 (d, C4 and C1), 123.56 (d, C3), 50.18 (d, -CHBr-CH3), 39.12 (t, C7), 33.80 (s, C8), 31.75 (q, C9 and C10 methyls), 30.44 (t, C5), 26.93 (q, -CHBr- CH3), and 19.52 (t, C6); m/z (%), 187 (M+-Br, C14H19, 100), 171 (12), 117 (19).
A mixture of the above bromide (0.772g, 2.9mmol) and PPh3 (0.866g, 3.3mmol) in 1.5mL xylene was refluxed at 144°C for 3 days. Addition of ether to the cooled reaction mixture followed by recrystallization from dichloromethane-toluene yielded 1.523g (100%) of phosphonium salt 14: mp 200-203 °C, IR 2190, 1592, 1440, 1115, 930, 730, 698 cm-1; 1H NMR 7.72 (m, -PPh3), 7.17 (s br, H-1), 6.71 (d br, J = 8, H-3), 6.83 (d, J = 8, H-4), 6.17 (sx, J = 6.8, -CHPPh3Br-CH3), 2.68 (t br, J = 7, H-5), 1.82 (dd, J = 7.2 and 19, -CHPPh3Br-CH3), 1.74 (m, H-6), 1.57 (m, H-7), 1.01 and 1.05 (s, C9 and C10 methyls).
(1E, 3E)- and (1E, 3Z)-m-[4-(5,6,7,8-Tetrahydro-8,8- dimethyl-2-naphthyl)-1,3-pentadienyl] phenols (15 and 16)
A mixture of phosphonium salt 14 (226mg, 0.427mmol), aldehyde 13 (114mg, 0.49mmol) and 15mL of 1,2-epoxybutane was refluxed at 70°C for 17h. The reaction mixture was cooled to room temperature, diluted with water and extracted with diethyl ether. The ether extract was washed with brine, dried and evaporated. Purification of the residue by preparative TLC on silica gel gave 75mg of THP protected retinoids, to which was added 8mL of ethyl acetate, 2mL of methanol, and 28mg of p-toluenesulfonic acid. After being stirred for 1h at room temperature, the reaction mixture was diluted with ether, washed with water, NaHCO3 solution and brine, dried and evaporated to give 59mg (44% yield) of E and Z retinoids 15 and 16 (85 : 15), which were separated by HPLC. Data for (1E, 3E)-m-[4-(5,6,7,8-tetrahydro- 8,8-dimethyl-2-naphthyl)-1,3-pentadienyl] phenol (15): mp 108-109 °C; IR 3320, 1600, 1582, 1450, 965 cm-1; 1H NMR 7.44 (d, J = 1.8, 1H), 7.17 (m, 3H), 7.01 (d, J = 8.1, 2H), 6.93 (d, J = 2.2, 1H). 6.68 (dd, J = 1.8 and 7.4, 1H), 6.58 (d br, J = 11 and 15, 2H), 5.08 (br, -OH), 2.76 (t, J = 6.3, 2H), 2.25 (s, 3H), 1.79 (m, 2H), 1.67 (m, 2H), and 1.32 (s, 6H); 13C NMR 155.60 (s), 145.55 (s), 140.43 (s), 139.60 (s), 137.56 (s), 135.41 (s), 131.71 (d), 129.71 (d), 128.98 (d), 126.34 (d), 126.15 (d), 123.71 (d), 122.69 (d), 119.17 (d), 114.35 (d), 112.69 (d), 39.31 (t), 33.90 (s), 31.80 (q, 2C), 30.44 (t), 19.61 (t), and 16.20 (q); UV 331 (36360), 238 (s), 216.5 (20130), 203 nm (23370); HRMS calc’d for C23H26O 318.1984, found 318.1988. Data for (1E, 3Z)-m- [4-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)-1,3-pentadienyl] phenol (16): IR 3390, 1600, 1580, 1450, 1160 cm-1; 1H NMR 7.25 (d, J = 1.8, 1H), 6.99 (m, 5H), 6.75 (d br, J = 2.2, 1H), 6.62 (dd, J = 2.2 and 8.1, 1H), 6.45 (d, J = 15.4, 1H), 6.27 (d, J = 11, 1H), 4.7 (br, -OH), 2.79 (t, J = 6.3, 2H), 2.18 (s, 3H), 1.82 (m, 2H), 1.69 (m, 2H), and 1.31 (s, 6H); 13C NMR 155.65 (s), 145.50 (s), 140.48 (s), 139.75 (s), 138.78 (s), 135.26 (s), 130.34 (d), 129.66 (d), 128.88 (d), 127.61 (d), 126.93 (d, 2C), 125.17 (d), 119.13 (d), 114.01 (d), 112.59 (d), 39.36 (t), 33.90 (s), 31.90 (q, 2C), 30.58 (t), 25.51 (q), and 19.76 (t); UV 310 (29690), 250 (s), 243 (14750), 211 (s), 201 nm (35720); HRMS calc’d for C23H26O 318.1984, found 318.1964.
Methyl (E)- and methyl (Z)-p-[2-(5,6,7,8-tetrahydro-8,8- dimethyl-2-naphthyl)propenyl] benzoates (18a and 19a)
A mixture of methyl p-formylbenzoate 17 (121mg, 0.737mmol), phosphonium salt 14 (332mg, 0.627mmol), and 20mL of 1,2-epoxybutane was refluxed at 70°C for 17h. The reaction mixture was cooled, diluted with water and extracted with diethyl ether. The ether extract was washed with brine, dried and evaporated. Purification of the residue by preparative TLC on silica gel gave 161mg (77% yield) of E and Z retinoid esters 18a and 19a in a ratio of 87:13. Irradiation of this mixture in dichloromethane using a Sylvania spotlight (150W) at reflux for 20h gave a 2:3 mixture of the esters, which were separated by HPLC. Data for methyl (E)-p-[2-(5,6,7,8- tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzoate (18a): mp 116-117 °C; IR 1722, 1613, 1225, 1112 cm-1; 1H NMR 8.03 (d, J = 8.4, 2H), 7.47 (d, J = 1.8, 1H), 7.43 (d, J = 8.5, 2H), 7.23 (dd, J = 2.2 and 7.7, 1H), 7.06 (d, J = 8.1, 1H), 6.79 (s br, 1H), 3.92 (s, 3H), 2.78 (t, J = 6.3, 2H), 2.29 (d, J = 1.1, 3H), 1.81 (m, 2H), 1.68 (m, 2H), and 1.33 (s, 6H); 13C NMR 167.01 (s), 145.75 (s), 143.26 (s), 141.16 (s), 139.90 (s), 135.80 (s), 129.41 (d, 2C), 129.02 (d, 3C), 127.76 (s), 125.90 (d), 124.20 (d), 123.08 (d), 51.94 (q), 39.31 (t), 34.00 (s), 31.85 (q, 2C), 30.39 (t), 19.61 (t), and 17.76 (q); UV 306 (26350), 233 (14120), 203 nm (29990); HRMS calc’d for C23H26O2 334.1933, found 334.1942. Data for methyl (Z)-p-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl) propenyl] benzoate (19a): IR 1720, 1600, 1272, 1105 cm-1; 1H NMR 7.77 (d, J = 8.1, 2H), 6.98 (m, 5H), 6.45 (s br, 1H), 3.83 (s, 3H), 2.73 (t, J = 6.3, 2H), 2.21 (d, J = 1.1, 3H), 1.77 (m, 2H), 1.60 (m, 2H), and 1.08 (s, 6H); 13C NMR 166.86 (s), 145.70 (s), 142.82 (s), 141.75 (s), 138.43 (s), 135.17 (s), 129.17 (d), 129.07 (d, 2C), 128.68 (d, 2C), 127.17 (s), 126.83 (d), 125.27 (d), 124.50 (d), 51.74 (q), 39.12 (t), 33.61 (s), 31.51 (q, 2C), 30.44 (t), 26.88 (q), and 19.61 (t); UV 298 (13660), 240 (s), 232 (15330), 203 nm (32230); HRMS calc’d for C23H26O2 334.1933 found 334.1927.
Ester 18a (28mg) was hydrolyzed by treatment with 10mL of 2N KOH (1.2g in 2mL water and 8 mL methanol) at 50°C (protected from light) for 3.5h. The reaction mixture was cooled, acidified with HCl and extracted with chloroform. The chloroform extract was washed with brine, dried and evaporated. Recrystallization of the residue from dichloromethane-hexane furnished 22 mg (82% yield) of (E)- p-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzoic acid (18b): mp 171-174 °C; IR 3170-2300, 1678, 1600, 1285 cm-1; 1H NMR 8.12 (d, J = 8.1, 2H), 7.47 ( s br, 1H), 7.46 (d, J = 8.1, 2H), 7.24 (dd, J = 1.8 and 8.1, 1H), 7.06 (d, J = 8.1, 1H), 6.81 ( s br, 1H), 2.78 (t, J = 6.3, 2H), 2.30 (d, J = 1.1, 3H), 1.82 (m, 2H), 1.69 (m, 2H), and 1.34 (s, 6H); 13C NMR 171.73 (s), 145.80 (s), 144.24 (s), 141.12 (s), 140.43 (s), 135.95 (s), 130.15 (d, 2C), 129.17 (d, 2C), 128.83 (d), 125.90 (d), 125.76 (s), 124.29 (d), 123.12 (d), 39.36 (t), 34.05 (s), 31.90 (q, 2C), 30.54 (t), 19.66 (t) and 17.86 (q); UV 293 (22140), 225 (s), 203 nm (34670); HRMS calc’d for C22H24O2 320.1776, found 320.1784.
Ester 19a (16mg) was similarly hydrolyzed by treatment with 10mL of 2N KOH (1.2g in 2mL water and 8mL methanol) at 50°C (protected from light) for 3h. The usual workup with chloroform after acidification with HCl gave 15mg (98% yield) of (Z)-p-[2-(5,6,7,8- tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzoic acid (19b): mp 174-177 °C; IR 3300-2300, 1682, 1600, 1290 cm-1; 1H NMR 7.83 (d, J = 8, 2H), 6.99 (m, 5H), 6.48 ( s br, 1H), 2.75 (t, J = 6.3, 2H), 2.23 (d, J = 1.1, 3H), 1.78 (m, 2H), 1.60 (m, 2H), and 1.08 (s, 6H); 13C NMR 172.17 (s), 145.89 (s), 143.89 (s), 142.38 (s), 138.43 (s), 135.41 (s), 129.80 (d, 2C), 129.32 (d), 128.88 (d, 2C), 126.98 (d), 126.39 (s), 125.27 (d), 124.54 (d), 39.22 (t), 33.75 (s), 31.56 (q, 2C), 30.54 (t), 26.98 (q), and 19.66 (t); UV 288 (11150), 237 (13490), 210 (s), 202 nm (28430); HRMS calc’d for C22H24O2 320.1776, found 320.1753.
(E)- and (Z)-p-[2-(5,6,7,8-Tetrahydro-8,8-dimethyl-2- naphthyl)propenyl] benzaldehydes (18d and 19d)
Ester 18a (28mg) in 10mL tetrahydrofuran was added to a slurry of LiAlH4 (70mg) in 5mL tetrahydrofuran and the mixture was stirred at 0°C for 1h. Workup in the usual manner gave 25mg (98% yield) of (E)-p-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzyl alcohol (18c): mp 68-70 °C; IR 3320, 1600, 1445, 1002, 808 cm- 1; 1H NMR 7.46 (d, J = 1.8, 1H), 7.37 (s br, 4H), 7.24 (dd, J = 1.8 and 7.7, 1H), 7.05 (d, J = 7.7, 1H), 6.77 (s br, 1H), 4.71 (s, 2H), 2.78 (t, J = 6.4, 2H), 2.27 (d, J = 1.1, 3H), 1.81 (m, 2H), 1.68 (m, 2H), and 1.33 (s, 6H); 13C NMR 145.65 (s), 141.51 (s), 138.82 (s), 138.00 (s, 2C), 135.36 (s), 129.32 (d, 2C), 128.98 (d), 126.83 (d, 2C), 126.44 (d), 124.15 (d), 123.08 (d), 65.15 (t), 39.41 (t), 34.00 (s), 31.90 (q, 2C), 30.49 (t), 19.71 (t), and 17.61 (q); UV 281 (22180), 225 (s), 208.5 nm (24300); HRMS calc’d for C22H26O 306.1984, found 306.1998.
Ester 19a (40mg) in 5mL tetrahydrofuran was added to a slurry of LiAlH4 (70mg) in 7mL tetrahydrofuran and the mixture was stirred at 0°C for 1.5h. Workup in the usual manner gave 36mg (98% yield) of (Z)-p-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzyl alcohol (19c): IR 3340, 1605, 1455, 1007, 865, 820 cm-1; 1H NMR 7.08 (m, 3H), 6.96 (m, 4H), 6.43 (s br, 1H), 4.54 (s, 2H), 2.73 (t, J = 6.3, 2H), 2.19 (d, J = 1.2, 3H), 1.77 (m, 2H), 1.61 (m, 2H), and 1.09 (s, 6H); 13C NMR 145.55 (s), 139.12 (s), 138.87 (s), 138.34 (s), 137.46 (s), 134.78 (s), 128.98 (d, 3C), 126.98 (d), 126.54 (d, 2C), 125.66 (d), 124.68 (d), 65.05 (t), 39.22 (t), 33.66 (s), 31.56 (q, 2C), 30.49 (t), 26.83 (q), and 19.66 (t); UV 276 (10650), 232.5 (3900), 208 (s), 203 (15460); HRMS calc’d for C22H26O 306.1984, found 306.1960.
A solution of alcohol 18c (28mg) in 10mL dichloromethane was oxidized by stirring with 140mg of activated MnO2 for 6h. The reaction mixture was filtered through a bed of silica gel, celite and activated charcoal, and washed with chloroform to give 27mg (97% yield) of (E)-p-[2-(5,6,7,8-tetrahyrdo-8,8-dimethyl-2-naphthyl) propenyl] benzaldehyde (18d): mp 116-118 °C; IR 1692, 1597, 1162 cm-1; 1H NMR 9.81 (s, 1H), 7.87 (d, J = 8.5, 2H), 7.51 (d, J = 8.1, 2H), 7.47 (d, J = 1.8, 1H), 7.23 (dd, J = 1.8 and 8.1, 1H), 7.06 (d, J = 8.1, 1H), 6.80 (s br, 1H), 2.78 (t, J = 6.3, 2H), 2.31 (d, J = 1.1, 3H), 1.81 (m, 2H), 1.68 (m, 2H), and 1.33 (s, 6H); 13C NMR 191.68 (d), 145.80 (s), 144.92 (s), 140.87 (s, 2C), 136.04 (s), 134.24 (s), 129.61 (d, 4C), 129.12 (d), 125.76 (d), 124.25 (d), 123.08 (d), 39.31 (t), 34.00 (s), 31.90 (q, 2C), 30.49 (t), 19.66 (t), and 17.91 (q); UV 321 (28490), 240 (15460), 204 (36030); HRMS calc’d for C22H24O 304.1827, found 304.1839.
A solution of alcohol 19c (24mg) in 12mL dichloromethane was oxidized by stirring with 120mg of activated MnO2 for 5h. Filtration of the reaction mixture through a bed of silica gel, celite, and activated charcoal, and washing with chloroform gave 22mg (93% yield) of (Z)-p-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] benzaldehyde (19d): mp 71-73 °C; IR 1692, 1600, 1162 cm-1; 1H NMR 9.87 (s, 1H), 7.61 (d, J = 8.5, 2H), 7.11 (d, J = 8.1, 2H), 7.05 (s br, 1H), 7.00 (d, J = 7.7, 1H), 6.92 (dd, J = 1.8 and 7.7, 1H), 6.47 (s br, 1H), 2.74 (t, J = 6.3, 2H), 2.23 (d, J = 1.1, 3H), 1.78 (m, 2H), 1.61 (m, 2H), and 1.06 (s, 6H); 13C NMR 191.73 (d), 145.89 (s), 144.72 (s), 142.97 (s), 138.34 (s), 135.56 (s), 133.95 (s), 129.41 (d, 5C), 126.98 (d), 125.22 (d), 124.54 (d), 39.17 (t), 33.75 (s), 31.56 (q, 2C), 30.54 (t), 27.02 (q), and 19.66 (t); UV 314.5 (16000), 243 (s), 234.5 (16020), 203 nm (34000); HRMS calc’d for C22H24O 304.1827, found 304.1819.
(E) and (Z)-3-Methyl-5-[2-(5,6,7,8-tetrahydro-8,8-dimethyl- 2-naphthyl)-propenyl] phenols (21 and 22)
A mixture of phosphonium salt 14 (237mg), 3-acetoxy-5- methylbenzaldehyde 20 (115mg), and 15mL of 1,2-epoxybutane was refluxed at 70°C for 14h. Dilution of the reaction mixture with water and workup in the usual manner with diethyl ether followed by preparative TLC on silica gel gave 75mg (49% yield) of a mixture of retinoid acetates, which was dissolved in 5mL tetrahydrofuran and added dropwise to a slurry of 159mg of LiAlH4 in 5mL tetrahydrofuran at 0°C. The reaction mixture was stirred at 0°C for 1h, quenched with EtOAc followed by water, and passed through silica gel to give 47mg (72% yield) of E and Z isomers 21 and 22 (85 : 15). The isomers were then separated by HPLC. Data for (E)-3-methyl-5-[2-(5,6,7,8- tetrahydro-8,8-dimethyl-2-naphthyl)propenyl] phenol (21): IR 3360, 1610, 1580, 1146 cm-1; 1H NMR 7.44 (d, J = 1.8, 1H), 7.19 (dd, J = 1.8 and 7.7, 1H), 7.01 (d, J = 7.7, 1H), 6.75 ( s br, 1H), 6.68 ( s br, 1H), 6.65 (s br, 1H), 6.52 (s br, 1H), 5.24 (br, -OH), 2.75 (t, J = 6.3, 2H), 2.29 (s, 3H), 2.24 (d, J = 1.1, 3H), 1.79 (m, 2H), 1.66 (m, 2H), and 1.31 (s, 6H); 13C NMR 155.11 (s), 145.60 (s), 141.51 (s), 139.95 (s), 139.31 (s), 138 (s), 135.26 (s), 128.93 (d), 126.44 (d), 124.15 (d), 123.03 (d), 122.73 (d), 114.15 (d), 112.98 (d), 39.36 (t), 33.95 (s), 31.85 (q, 2C), 30.44 (t), 21.32 (q), 19.66 (t), and 17.66 (q); UV 280 (18460), 215 (27170), 203 nm (s); HRMS calc’d for C22H26O 306.1984, found 304.1996. Data for (Z)-3-methyl-5-[2-(5,6,7,8-tetrahydro-8,8-dimethyl-2-naphthyl) propenyl] phenol (22): IR 3400, 1605, 1580, 1145 cm-1; 1H NMR 7.11 (s br, 1H), 6.99 (d, J = 8, 1H), 6.94 (dd, J = 1.8 and 8, 1H), 6.41 (s br, 1H), 6.37 (s br, 1H), 6.34 (s br, 1H), 6.21 (s br, 1H), 4.33 (br, -OH), 2.74 (t, J = 6.3, 2H), 2.17 (d, J = 1.5, 3H), 2.13 (s, 3H), 1.77 (m, 2H), 1.61 (m, 2H), and 1.10 (s, 6H); 13C NMR 154.91 (s), 145.50 (s), 141.80 (s), 139.31 (s), 139.12 (s), 138.97 (s), 134.83 (s), 129.02 (d), 127.17 (d), 125.76 (d), 124.59 (d), 122.69 (d), 113.76 (d), 112.50 (d), 39.31 (t), 33.70 (s), 31.51 (q, 2C), 30.54 (t), 26.83 (q), 21.27 (q), and 19.76 (t); UV 272 (8210), 230 (s), 213 (s), 203 (35900); HRMS calc’d for C22H26O 306.1984, found 304.1965.
TOC assay of the new retinoids
Biological activity of all new synthetic retinoids was evaluated using the hamster tracheal organ culture (TOC) assay reported by Newton, Henderson and Sporn [15]. Tracheas from hamsters in early stages of vitamin A deficiency were placed in organ culture and maintained in a medium containing no retinoid for 3 days. They were then treated with retinoids dissolved in DMSO. For retinoids 10a, 10b, and 18a, 54-60 cultures were used. For retinoids 15, 18b, and 18d, 35-41 cultures were used. For all other retinoids, 20-26 cultures were used. After 10 days, cross sections of the mid portions were examined with a microscope for the presence of keratin and keratohyaline granules. An observation was scored as ‘active’ if neither keratin nor keratohyaline granules were seen, or if keratohyaline granules alone were absent. An observation was scored as ‘inactive’ if both keratin and keratohyaline granules were seen. Dose-response curves were made for the reference substance “all-trans-retinoic acid” and each new retinoid, and from these data 50% effective dose ED50(M) was determined.
Conclusion
In all of the fifteen new synthetic retinoids, 9-trans retinoids (E isomers) are more active than their 9-cis retinoids (Z isomers). Comparing the ED50(M) values of the most active retinoids, the 9E isomers are 40-60 times more active than the corresponding 9Z isomers. In conclusion, we developed a new series of retinoids, three of which (18a, 18b, and 10b) exhibit ED50(M) values in the (3-5)x10-11 range, and are promising candidates for the prevention and treatment of epithelial cancer.
Acknowledgement
This work was supported by the National Cancer Institute through contract No. 1-CP-05716.
References
- The work in this paper was performed by Mohanraj SM at Cornell University, NY.
- Alvarez R, Vaz B, Gronemeyer H, Lera AR. Functions, therapeutic applications, and synthesis of retinoids and carotenoids. Chem. Rev. 2014; 114: 1-125.
- Orfanos CE, Braun-Falco O, Farber EM, Grupper C, Polano MK, Schuppli R. eds. Retinoids. Springer-Verlag, Berlin. 1981.
- Moon RC, Itri LM. In Sporn MB, Roberts AB, Goodman DS. eds. The Retinoids, Vol. 2, Academic Press, Orlando.1984; 327-372.
- Peck GL. in Sporn MB, Roberts AB, Goodman DS. eds. The Retinoids, Vol. 2, Academic Press, Orlando. 1984; 391-412.
- Dennert G. in Sporn MB, Roberts AB, Goodman DS. eds. The Retinoids, Vol. 2, Academic Press, Orlando. 1984; 373-390.
- Kamm JJ, Ashenfelter KO, Ehmann CW. in Sporn MB, Roberts AB, Goodman DS. eds. The Retinoids, Vol. 2, Academic Press, Orlando.1984; 288-326.
- (a) Loelinger P, Bollag W, Mayer H. Eur. J. Med. Chem. Chim. Ther. 1980; 15: 9-15; (b) Dawson, M. I.; Hobbs, P. D.; Derdzinski, K. A.; Chao, W. R.; Frenking, G.; Loew, G. H.; Jetten, A. M.; Napoli, J. L.; Williams, J. B. J. Med. Chem. 1989; 32:1504-1517.
- McMurry JE, Scott WJ. A method for the regiospecific synthesis of enol triflates by enolate trapping. Tetrahedron Lett. 1983; 24: 979-982.
- Boeckman RK, Bruza KJ. Chemistry of diorganocuprates containing functionalized ligands. 2. Methodology for conjugate addition of synthetic equivalents of enolates and acyl anions. J. Org. Chem.1979; 44: 4781-4788.
- McMurry JE, Scott WJ. A new method of olefin synthesis. Coupling of lithium dialkylcuprates with enol triflates. Tetrahedron Lett. 1980; 21: 4313-4316.
- Davalian D, Heathcock CH. Synthesis of 7,8-epoxy-7,8-dihydroretinoids. J. Org. Chem. 1979; 44: 4458-4461.
- Mohanraj S. J. High Resoln. Chromatog & Chromatog. Commun. 1984; 7: 46.
- Mohanraj S. Semi-Preparative HPLC Separations of E and Z Isomers of New Aromatic Retinoids. J. Liquid Chromatog. 1984; 7: 1455-1460.
- Newton DL, Henderson WR, Sporn MB. Structure-Activity Relationships of Retinoids in Hamster Tracheal Organ Culture. Cancer Research. 1980; 40: 3413-3425.
- We thank Merritt JE, Senior Associate Editor, Nomenclature, Chemical Abstract Service, Columbus, OH, for her assistance in naming and numbering the new retinoids.