
Research Article
Austin J Anat. 2021; 8(2): 1101.
Valproic Acid (VPA) in Combination with Knockdown of AKT3 and PI3KCA Genes Inhibits Proliferation, Induces Apoptosis and Autophagy in T98G and U87MG Glioblastoma Multiforme Cells
Paul-Samojedny M*, Liduk E, Borkowska P, Kowalczyk M, Suchanek-Raif R, Zielinska A and Kowalski J
Department of Medical Genetics, School of Pharmacy in Sosnowiec, Medical University of Silesia, Katowice, Poland
*Corresponding author: Paul-Samojedny M, Department of Medical Genetics, School of Pharmacy in Sosnowiec, Medical University of Silesia in Katowice, Jednosci 8 Street, 41-200 Sosnowiec, Poland
Received: July 13, 2021; Accepted: July 27, 2021; Published: August 03, 2021
Abstract
Purpose: Glioblastoma Multiforme (GBM) is a heterogenous and highly vascularized brain tumor that avoid apoptosis due to P-glycoprotein (P-gp) mediated multi-drug resistance. Therefore, development of new therapeutic strategies that induce apoptosis, inhibit proliferation, and overcome multi-drug resistance is urgently warranted. We examined the efficacy of combination of Valproic Acid (VPA) and knockdown of AKT3 and PI3KCA genes in human glioblastoma T98G and U87MG cell lines.
Material and Methods: T98G and U87MG cells were transfected with AKT3 or PI3KCA siRNAs. Transfection efficiency was assessed using Flow Cytometry (FC) and fluorescence microscopy. The influence of AKT3 and PI3KCA siRNAs in combination with VPA on T98G and U87MG cell viability, proliferation, apoptosis and autophagy was evaluated as well. Alterations in the mRNA expression of apoptosis-related genes (CASP3 and Bid) were analyzed using QRT-PCR.
Results: The transfection of T98G and U87MG cells with AKT3 or PI3KCA siRNAs and exposition on VPA led to a significant reduction in cell viability, the accumulation of subG1-phase cells and a reduced fraction of cells in the S and G2/M phases, apoptosis or necrosis induction and induction of autophagy.
Conclusions: The siRNA-induced AKT3 and PI3KCA mRNA knockdown in combination with VPA may offer a novel therapeutic strategy to more effective control the growth of human GBM cells. Thus, knockdown of these genes in combination with valproic acid inhibits proliferation, induces apoptosis and autophagy in T98G and U87MG cells, but further studies are necessary to confirm a positive phenomenon for the treatment of GBM.
Keywords: Glioblastoma multiforme; Valproic acid; siRNA; Proliferation; Cell death
Introduction
Valproic Acid (2-propylpentanoic acid; VPA) is a branched short-chain fatty acid used as an antiepileptic drug traditionally used for the treatment of certain types of seizures [1,2]. Meta-analysis performed by Yuan et al. suggests that glioblastoma patients may experience prolonged survival due to VPA administration [3]. For being identified as a potent selective histone deacetylase inhibitor, VPA has gained much attention of its potential central role in epigenetic gene regulation. Histone lysine residues acetylation and deacetylation are posttranslational modifications that influence gene expression activating and repressing gene transcription, respectively [1]. Thus, histone deacetylases play an important role in epigenetic regulation of various programmed cellular processes both in physiological and pathological conditions. VPA is often used to treat seizures in glioblastoma patients. It is known that VPA inhibits cell proliferation by causing cell-cycle arrest in the G1 and/or G2 phase and that it induces differentiation and/or apoptosis in cancer cells. There are evidences that VPA also reduced proliferation rates in glioblastoma-derived stem cells and decreased cell viability of primary human glioblastoma cells. It is very important that VPA may downregulate the expression of MGMT (O-6-methylguanine-DNA methyltransferase) and sensitize human glioma cells to temozolomide and irradiation [4]. Valproic acid has been also shown to induce cellular differentiation, growth arrest and apoptosis in gliomas [5].
Malignant gliomas are characterized by lack of response to implemented therapy leading to a quick recurrence and short lifetime, as well as chemo- and radiotherapy resistance. One of the reasons of observed clinical therapy resistance and malignance of human gliomas is overexpression of PI3K/Akt pathway molecules. PI3Ks (phosphoinositide 3-kinases) constitute a family of lipid kinases that are capable of phosphorylating the 3’OH of the inositol ring in phosphoinositides. These kinases are divided into three classes - class I consists of two subclasses-class IA and class IB, respectively. Class IA includes heterodimers that are composed of a p110 catalytic subunit and a p85 regulatory subunit. A p110 subunit has three isoforms (p110a, p110β and p110γ) that are encoded by the three different genes [6-8] and these isoforms are involved in the regulation of processes such as proliferation, cell survival, degranulation, vesicular trafficking and cell migration.
It is known that in cells in which the p110a isoform of PI3K is predominant or in which both p110a and p110β isoforms are equally important, the knockdown of PIK3CA (p110a) interferes with PI3K/ AKT signaling [9]. The PI3KCA gene has been found to be amplified and overexpressed in several types of cancers including gliomas. It has been suggested that the point mutations that activate the PI3KCA gene may represent a novel mechanism for the induction oncogenic PI3K signaling pathway [10,11]. The PI3K gene amplification may cause the increased AKT activity in tumors. The activation of AKT has been found in approximately 80% of human GBMs [12].
The AKT kinase plays an important role in the PI3K signaling pathway and AKT activity is induced following PI3K activation in various growth factor receptor-mediated signaling cascades [13]. There are three isoforms of AKT (AKT1, AKT2 and AKT3), which are encoded by three different genes. The AKT2 and AKT3 (but not AKT1) isoforms are pathologically amplified in human cancers [14]. AKT3 is primarily expressed in the brain and testis [15]. It is known that AKT2 and AKT3 are overexpressed in glioma cells and play a pivotal role in malignant gliomas [16]. Bearing in mind that cellular behavior is result of intracellular signaling network activation and depends on cross talks between different pathways, it is very interesting to verify if siRNA silencing of PI3K/Akt pathway accelerate VPA-mediated effects.
Thus, the aim of our study is concerned on proliferation, apoptosis and autophagy processes induction and changes in cell cycle AKT3 and PIK3CA genes knockdown and valproic acid exposure. Current study was also undertaken to examine the effect of the siRNAs targeting AKT3 and PIK3CA genes on T98G cells susceptibility on VPA.
Materials and Methods
Cell cultures
The T98G (American Type Culture Collection - ATCC, Manassas, VA, USA) cell line derived from a 61-year-old male [17] and U87MG (Sigma Aldrich) cell line derived from a 44-year-old male. Glioblastoma multiforme cells were cultured in modified Eagle’s Minimum Essential Medium (ATCC) supplemented with heatinactivated 10% fetal bovine serum (ATCC) and 10μg/ml gentamicin (Invitrogen). Cell lines was maintained at 37°C in a humidified atmosphere of 5% CO2 in air.
Knockdown of AKT3 and PI3KCA genes using specific siRNA
Transfection of T98G and U87MG cells with specific siRNAs targeting AKT3 or PI3KCA mRNA was performed using FlexiTube siRNA Premix (Qiagen, Italy) according to the manufacturer’s protocol, as we have described previously [18].
Valproic acid uptake
Freshly prepared stock solutions of VPA was made in serum-free medium just prior to treatment. Dose-response studies were carried out to determine the suitable doses of the drug for the inhibition of cell growth and induction of cell death and 0.5mM concentration of VPA was chosen. T98G and U87MG GBM cells transfected with AKT3 and PI3KCA specific RNAs were treated with valproic acid (0.5mM) for 96h (48h after transfection).
Cell cycle analysis
T98G an U87MG cells were seeded in 6-well plates (at a density of 1.6 x 104 cells per well), cultured overnight (24h) and after AKT3 and PI3KCA genes siRNA silencing, cells were exposure to valproic acid and cell cycle was analyzed. After incubation with specific siRNA, cells were washed with PBS buffer, trypsinized and washed once more with cold PBS buffer. Then, after suspending the cells in 300μl of cold PBS buffer, 70% cold ethanol was added dropwise in a volume of 4mL on a vortex. The cell suspension in ethyl alcohol was incubated at -20°C for 1h, and the contents of the tube were gently mixed every 10 minutes. After incubation with ethanol, the fixed cells were centrifuged and washed with PBS buffer. The cells were then resuspended in 250μl of PBS and RNaseA (10mg/ml final concentration) and propidium iodide (1mg/ml final concentration) were added. Cells were incubated for 1h under standard conditions, and the contents of the tube were gently mixed every 10 minutes. The percentage of cells at different phases of the cell cycle (subG1, G1/G0, S, G2/M, polyploidy) was assessed using a FACSAria II flow cytometer (Becton Dickinson, USA).
Apoptosis assay
T98G and U87MG cells were seeded in 6-well plates (at a density of 1.6 x 104 cells per well), cultured overnight (24h) and after AKT3 and PI3KCA genes siRNA silencing, cells were treated with valproic acid. Cells were analyzed using flow cytometry and a Vybrant® DyeCycleTM Violet/SYTOX® AADvancedTM Apoptosis Kit as described previously [18].
Total RNA extraction
Total RNA was isolated from cells cultured by TRIzol reagent (Life Technologies, Inc, Grand Island, NY, USA) according to the manufacturer’s protocol. The integrity of total RNA was checked by electrophoresis in 1% agarose gel stained with ethidium bromide. All RNA extracts were treated with DNAse I to avoid genomic DNA contamination, and assessed qualitatively and quantitatively.
Changes in mRNA copy number of apoptosis-related genes by RT-qPCR
We have performed QRT-PCR for selected genes associated with apoptosis: CASP3 and BID. RT-qPCR assays were performed using specific primers (KiCqStartTM Primers; Sigma Aldrich) and SensiFASTTM SYBR Hi-ROX One-Step kit (Bioline) according to the manufacturer’s protocol. (CFX96 Real-Time System; BIO-RAD). Real-time fluorescent RT-PCR was performed using Following conditions were taken: 45°C for 10min, 95°C for 2min, followed by 40 cycles of 5sec at 95°C, 10sec at 60°C and 5sec at 72°C. RNA for human TBP (Tata Binding Protein) was used as an endogenous control. The copy numbers for each sample were calculated by the CT-based calibrated standard curve method. Each of 12 data point for mRNA copy numbers is the average of duplicates on the same analyzed plate.
Autophagy identification using LysoTracker Red and flow cytometry
T98G and U87MG cells with or without treatment were loaded LysoTracker Red (LTR) (100nM; Invitrogen), according to the manufacturer’s instructions by incubating cells with dye for 15min at 37°C, respectively. LTR loading was shown to be optimal at 100nM after 15 minutes incubation for both cell lines. Cells were then washed in PBS buffer and resuspended in 500μl of PBS. Cells were analyzed for LTR Median Fluorescence Intensity (MFI) levels by first gating on all cell material except small debris in the origin of a FSC versus SSC dot-plot.
Statistical analysis
Data were presented as mean ± SD. One way ANOVA and post hoc Tukey’s multiple comparison test were used for comparing analyzed groups. The power of all tests was not less than 1-β=0.8. Data were analyzed with the Statistica software (StatSoft, Inc. 2008), version 10.0 (www.statsoft.com). All of the tests were two-sided and p<0.05 was considered to be statistically significant. The proliferation and apoptotic indexes were determined according to Darzynkiewicz et al. (1994) and Henry et al. (2008) [19,20]. Hierarchical clustering of the results based on the Euclidean distance was carried out using GenExEnterprise 5.4.3.703. QRT-PCR analysis was performed in accordance with the mathematical rules that this program uses. In order to identify differentially expressed, autophagy- and apoptosisrelated genes, linear regression was performed. Data of QRT-PCR analysis were also clustered using SOMs.
Results
Cell cycle changes after siRNA silencing and valproic acid treatment
In order to examine the possible mechanisms of the antiproliferative activity of AKT3 and PI3KCA siRNAs combination with Valproic Acid (VPA), cell cycle distribution using flow cytometry was performed. Proliferation index (PI), i.e. percentage of proliferating cells in S+G2/M cell cycle phases, was determined. PI was quantified in untransfected T98G and U87MG cells and after the knockdown with adequate 1nM siRNA and valproic acid exposition. It was found that AKT3 and PI3KCA siRNAs increase the percentage of the cells in the subG1 phase as compared to untransfected (control) T98G (8.4% vs. 41.1% and 52%) and U87MG cells (1.6% vs. 20.7% and 18.7%) (Figure 1A and 1B) and AKT3 and PI3KCA siRNAs in combination with 0.5mM valproic acid decreased percentage of the T98G (Figure 1A) (14% vs. 5.1% and 5% for S phase; 5.7% vs. 2.2% and 1.9% for G2/M phase) and U87MG (Figure 1B) (15.1% vs. 10.4% and 9.6% for S phase; 13.2% vs. 5.6% and 8.6% for G2/M phase) cells in the G2/M and S phases as compared to untransfected (control) after their exposition to VPA.
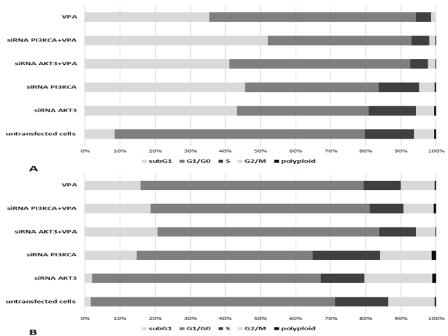
Figure 1: The cell cycle distribution in the T98G (A) and U87MG (B) cells that had been transfected with AKT3 and PI3KCA siRNAs and treated with Valproic Acid
(VPA), respectively. Results are presented as a percentage contribution of number of cells located in each cell cycle phase, including subG1 population. Presented
data are obtained from DNA histograms and represent an average of six independent repeats.
We have also revealed that AKT3 and PI3KCA siRNAs in combination with 0.5mM VPA decrease proliferation index, as compared to untransfected (control) T98G (17.68% vs. 7.03% and 6.9%) and U87MG (28.12% vs. 15.75% and 18.15%) cells after or no their exposition to VPA. It was found that AKT3 and PI3KCA siRNAs increases proliferation index as compared to untransfected (control) U87MG cells (Figure 2).
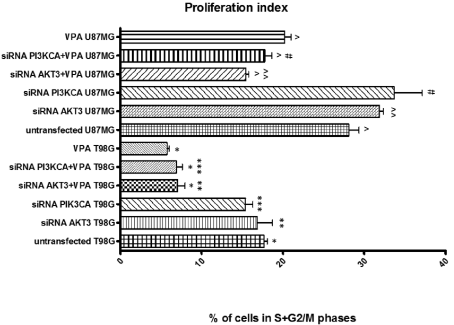
Figure 2: The proliferation index in the T98G and U87MG cell culture that had been transfected with AKT3 and PI3KCA siRNAs and treated with Valproic Acid
(VPA), respectively. Presented data are obtained from DNA histograms and represent an average of six independent repeats. Bar graph shows the mean of the
percentage of cell in S and G2/M phases with error bars (S.E.M.) from three independent experiments. To estimate statistical significance date were analyzed by
one way ANOVA and post hoc Tukey’s multiple comparison test and Statistica software (StatSoft, Inc. 2008), version 10.0 (www.statsoft.com) (p <0.05); *, **, ***,
^, ^^, #: Indicates a significant difference between analyzed groups.
Apoptosis and necrosis induction after siRNA silencing and valproic acid treatment
To further characterize glioblastoma cell response to siRNAs and VPA, we next analyzed apoptosis by flow cytometry. Necrotic and apoptotic cells were detected using flow cytometry and double staining with mentioned apoptosis kit following siRNA silencing and valproic acid exposition. The knockdown of AKT3 and PI3KCA genes in combination with VPA led to apoptosis induction in 43.8% cells (AKT3 siRNA+VPA) and 45.9% cells (PI3KCA siRNA+VPA), respectively, compared to 8.7% in the untransfected (control) T98G cells and in 54.9.8% cells (AKT3 siRNA+VPA) and 69.9% cells (PI3KCA siRNA+VPA), respectively, compared to 33.47% in the untransfected (control) U87MG cells (Figure 3A and 3B; p<0.05; by one way ANOVA and post hoc Tukey’s multiple comparison test).
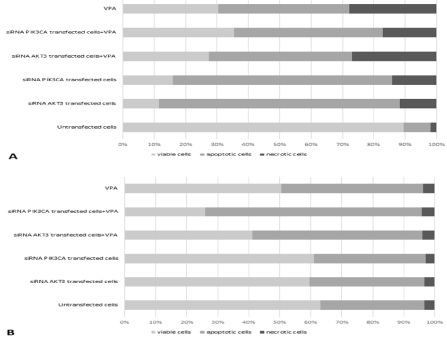
Figure 3: The influence of AKT3 and PI3KCA genes knockdown on the induction of apoptosis and necrosis in T98G and apoptosis in U87MG cells treated with
Valproic Acid (VPA). Results are presented as a percentage contribution (Y-axis) of number of viable cells, apoptotic cells and necrotic cells (A) in the groups that
were analyzed. The data are expressed as the means of four separate experiments (p <0.05; by one-way ANOVA and post hoc Tukey’s multiple comparison test).
In contrast, the necrosis rates of transfected T98G cells were 25.9% and 16.6% after AKT3 and PI3KCA silencing in combination with VPA, respectively, compared to the necrosis rate of the untransfected (control) cells, which was only 1.8%. (Figure 3A; p <0.05; by oneway ANOVA and post hoc Tukey’s multiple comparison test). We observed a 5-fold and 5.3-fold higher apoptotic index value after cell transfection with AKT3 and PI3KCA siRNAs in combination with VPA for T98G cells (Figure 3A; p<0.05; by one way ANOVA and post hoc Tukey’s multiple comparison test) and 1.6-fold and 2.1-fold higher apoptotic index value after cell transfection with AKT3 and PI3KCA siRNAs in combination with VPA for U87G cells (Figure 3B; p<0.05; by one way ANOVA and post hoc Tukey’s multiple comparison test).
We found that the changes in apoptosis-related genes (CASP3 and Bid) after AKT3 knockdown in combination with VPA are manifested mainly in: (1) decreased CASP3 gene expression in U87MG cells as well as increased CASP3 gene expression in T98G cell. The CASP3 gene expression was highest in T98 cells after PI3KCA knockdown in combination with VPA (Figure 4A and 4B).
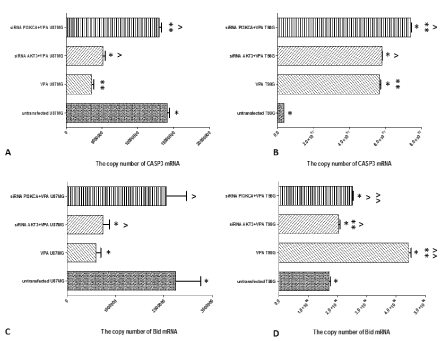
Figure 4: A comparison of mRNA copy number (X-axis) of apoptosis-related genes (CASP3 and Bid) differentiating cells transfected with AKT3 and PI3KCA
siRNAs and treated with Valproic Acid (VPA) from untransfected cells. A, B) The copy number of CASP3 mRNA (A - In U87MG cells, B - In T98G cells); C, D)
The copy number of Bid mRNA (C - In U87MG cells, D - In T98G cells). To estimate statistical significance (B) date were analyzed by one way ANOVA and post
hoc Tukey’s multiple comparison test and Statistica software (StatSoft, Inc. 2008), version 10.0 (www.statsoft.com) (p <0.05). *, **, ^, ^^: Indicates a significant
difference between analyzed groups.
Silencing of the AKT3 gene in combination with VPA is connected with decreased Bid gene expression in U87MG cells, as well as silencing of the AKT3 and PI3KCA genes in combination with VPA is connected increased Bid gene expression in T98G cells (Figure 4C and 4D).
Autophagy induction in T98G and U87MG GBM cells with down-regulated expression of AKT3 and PI3KCA genes exposed to valproic acid
To test our hypothesis that the knockdown of AKT3 or PI3KCA gene in cells exposed to VPA causes the induction of autophagy, we tested it with LysoTracker Red (a deep red-fluorescent dye for labeling and tracking acidic organelles in live cells) that preferentially accumulates in vesicles with an acidic pH and may be used to examine the efficiency of autophagosome/lysosome fusion in live cells. Our results indicate that AKT3 and PI3KCA genes silencing in T98G and U87MG cells exposed to VPA is associated with an increased intensity of red fluorescence, which indicates an increased number of vesicles with an acidic pH characteristic for autophagy (Figure 5; *^ p <0.05; by one way ANOVA and post hoc Tukey’s multiple comparison test). These changes are more significant in U87MG than in T98G cells.
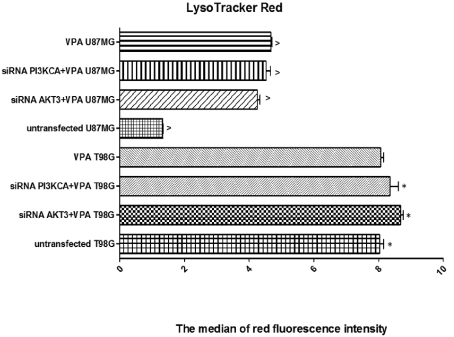
Figure 5: A comparison of red fluorescence intensity (after LysoTracker Red staining) indicated an increased number of vesicles with an acidic pH characteristic
for autophagy in cells that had been transfected with a specific siRNAs as well untransfected T98G and U87MG cells exposed to Valproic Acid (VPA). The red
fluorescence intensity after LysoTracker Red staining was determined using flow cytometry; (p<0.05; by one-way ANOVA and post hoc Tukey’s multiple comparison
test); * and ^: Indicates a significant difference between untransfected cells and analyzed groups.
Discussion
Numerous studies have shown promising results with Histone Deacetylase (HDAC) inhibitors in various malignancies also including brain cancers [21]. Actually new therapeutic strategies are being tested in patients with GBM, such as HDAC inhibitor (HDACi) either alone or in combination with other therapies. HDACi activity inhibition on class I and IIa HDACs has been described [22-24], and anti-neoplastic effects have been demonstrated in multiple preclinical studies in various solid tumours including glioma [25-28].
It is known, that aberrant or overexpressed Akt signaling is a major event in glioblastoma, therefore our aim was to characterize the molecular changes induced by HDAC inhibitor - valproic acid (VPA) that are used for T98G and U87MG glioblastoma cells after knockdown of AKT3 and PI3KCA genes.
Glioblastoma multiforme (GBM) is most common brain cancer with high aggressiveness and resistance to conventional anti-tumoral therapies. Despite optimal multimodality treatment including debulking surgery, radiotherapy and temozolomide chemotherapy, the median survival is 12–15 months [21].
Two HDACi - Suberanilohydroxamic acid (SAHA) and valproic acid (VPA) has been tested by Cornago et al. (2014) to characterization their effects on glioma cell growth in vitro and to determine the molecular changes that promote cancer cell death. We have analyzed influence of VPA in combination with knockdown of AKT3 and PI3KCA genes on T98G and U87MG GBM cells. We found that both this combine method change cell cycle progression, reduce glioblastoma cell viability, proliferation and induce apoptosis and autophagy.
According to Cornago et al. (2014) we found decrease percentage of the cells in the G2/M phase after VPA treatment, but this effect was higher in combination with AKT3 and PI3KCA specific siRNAs. Cornago et al. (2014) found that HDACi alter cell cycle progression by decreasing the expression of G2 checkpoint kinases Wee1 and checkpoint kinase 1 (Chk1) and reduce the expression of proteins involved in DNA repair (Rad51), mitotic spindle formation (TPX2) and chromosome segregation (Survivin) in glioma cells and in human glioblastoma multiforme primary cultures [4]. Contrary to us earlier studies showing that, VPA in LN18 and U251MG cell lines caused G2/M arrest [29]. Our findings showed that VPA in combination with knockdown of AKT3 and PI3KCA genes in T98G and U87MG cell lines caused G1/G0 arrest. Similar to us other studies reported that VPA induces not only growth arrest and apoptosis, but also senescence in medulloblastoma and glioma cell lines by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and c-Myc [29-31].
It is known, that HDAC inhibitors typically inhibit cancer cell proliferation by induction of cell cycle arrest, differentiation and/or apoptosis. Our results revealed that VPA in combination with AKT3 and PI3KCA siRNAs decrease proliferation index. Bacon et al. (1997) showed the growth inhibitory effect of VPA upon C6 glioma cultured continuously in the presence of 1mM valproate. Authors suggested, that it is associated with an inhibition of transient alpha2,3 sialylation of a 65 kDa glycoprotein expressed maximally at 4h into the G1 phase of the cell cycle [32]. VPA at the same concentration has also shown profound growth inhibitory activity of D54 glioma cell line [26]. Knüpfer et al. (1998) revealed that VPA at concentrations ranging from 0.1 to 1 mM strongly inhibits proliferation of A172, 86HG39, 85HG66 and rat glioma C6 cells in a dose-dependent manner, whereas T98G cell growth remained unchanged [33].
Cell cycle analysis showed that combination therapy (VPA and AKT3 and PI3KCA specific siRNAs) increased the apoptotic subG1 population and apoptosis was further confirmed by flow cytometry and RT-QPCR. Combination therapy has also caused increase of caspase-3 (CASP3) and Bid mRNA copies in T98G cells. This result may indicate the induction of apoptosis. There is evidence that all HDAC inhibitors activate either one or both (extrinsic and intrinsic) pathways of apoptosis in cancer models [5,34].
Das et al. (2007) reported that VPA reduced cell proliferation, induced cytotoxicity in human glioblastoma cells, and increased GFAP expression for astrocytic differentiation [29]. Our results show that the knockdown of AKT3 or PI3KCA gene in cells exposed to VPA causes the induction of apoptosis of T98G and U87MG cells. In addition, when VPA and specific siRNAs were combined an additive, rather than synergistic effect was noted. Hosein et al. (2015) analyzed the effect of valproic acid in combination with irradiation and temozolomide on primary human glioblastoma cells and they found synergistic effect of this combination [35].
Our study revealed that the transfection of T98G and U87MG cells with AKT3 or PI3KCA siRNAs and exposition on VPA led to a significant induction of necrosis in T98G cells. Similar to us Schwartz et al. (2007) reported that VPA induces nonapoptotic cell death in multiple myeloma cell lines [36]. Bollino et al. (2015) report that VPA induces neuronal cell death through an atypical calpain-dependent necroptosis pathway connected with downstream activation of c-Jun N-terminal kinase 1 (JNK1) and increased expression of receptorinteracting protein 1 (RIP-1). Authors suggest, that it is accompanied by cleavage and mitochondrial release/nuclear translocation of apoptosis-inducing factor, mitochondrial release of Smac/DIABLO, and inhibition of the anti-apoptotic protein X-linked inhibitor of apoptosis (XIAP). Results indicate that VPA induces phosphorylation of the necroptosis-associated histone H2A family member H2AX, which is known to contribute to lethal DNA degradation. There is evidence that these signals are inhibited in neuronal cells that express constitutively activated MEK/ERK and/or PI3-K/Akt survival pathways [37].
Previous studies indicated that several anticancer drugs have been shown to induce not only apoptosis, but also autophagy in cancer cells [38]. There is evidence that autophagy can have a tumor-suppressive role, and loss of autophagy regulators has been described in several human cancers [39]. Autophagy may be important in the regulation of cancer development as well as progression and in determining the response of tumor cells to anticancer therapy. Therefore, we have analyzed efficacy of VPA in combination with silencing of AKT3 and PI3KCA genes on this type of cell death. A few reports suggest that HDAC inhibitors, including VPA, induce autophagy in mammalian cells [40]. Ouyang et al. (2013) showed that valproic acid induced conversion of LC3-I to LC3-II and formation of LC3 puncta, the typical markers of autophagy, in prostate cancer LNCaP and PC-3 cells [41]. Accordance to these observations we showed induction of autophagy by VPA, but this process was more pronounced after knockdown of AKT3 and PI3KCA genes and exposition to VPA. Similar to previous study [40], we have found that even in apoptosisproficient U87MG cells that harbor wild-type P53 and functional caspase-3, most of the cell death can be attributed to autophagy. Fu et al. (2010) showed that VPA induced autophagy in malignant glioma cells through modulating oxidative stress [40].
It should be taken into account that effects of VPA seem to be cell type specific, which may depend also on the level of differentiation and the underlying genetic alterations [25].
Conclusions
In conclusion, our results show that VPA in combination with knockdown of AKT3 and PI3KCA genes is more efficient than VPA in killing glioblastoma cells, reducing proliferation and inducing autophagy. Therefore, combination of specific siRNAs and VPA can be used as an effective treatment for controlling growth of human glioblastoma cells, but further studies are urgently needed to fully elucidate the mechanism behind these phenomena.
Declaration
Compliance with Ethical Standards: This work was supported by the grant from Medical University of Silesia. The University had no further role in study design, in the collection, analysis and interpretation of data, in the writing of the report nor in the decision to submit the paper for publication. Award Number: KNW-1- 128/N/4/0 Recipient: Jan Kowalski, Prof.
Informed consent was obtained from all individual participants included in the study.
Role of Funding Source: This work was supported by the grant from Medical University of Silesia (KNW-1-088/N/8/0). The University had no further role in study design, in the collection, analysis and interpretation of data, in the writing of the report nor in the decision to submit the paper for publication.
References
- Činčárová L, Zdráhal Z, Fajkus J. New perspectives of valproic acid in clinical practice. Expert Opin Investig Drugs. 2013; 22: 1535-1547.
- Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013; 46: 1323-1338.
- Yuan Y, Xiang W, Qing M, Yanhui L, Jiewen L, Yunhe M. Survival analysis for valproic acid use in adult glioblastoma multiforme: a meta-analysis of individual patient data and a systematic review. Seizure. 2014; 23: 830-835.
- Hoja S, Schulze M, Rehli M, Proescholdt M, Herold-Mende C, Hau P, et al. Molecular dissection of the valproic acid effects on glioma cells. Oncotarget. 2016; 7: 62989-63002.
- Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, et al. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014; 5: e1435.
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8: 627-644.
- Jiang B-H, Liu L-Z. PI3k/PTEN signaling in tumorigenesis and angiogenesis. Biochimica et Biophysica Acta. 2008; 1784: 150-158.
- Geering BCP, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA. 2007; 104: 7809-7814.
- Sternberger M, Schmiedeknecht A, Kretschmer A, Gebhardt F, Leenders F, Czauderna F, et al. GeneBlocs are powerful tools to study and delineate signal transduction processes which regulate cell growth and transformation. Antisense Nucleic Acid Drug Dev. 2002; 12: 131-143.
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304: 554.
- Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drugresistance. Int J Oncol. 2012; 40: 639-644.
- Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell. Signal. 2009; 21: 470-476.
- Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, et al. Up-regulation of AKT3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. Biol. Chem. 1999; 274: 21528- 21532.
- Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3’ kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003; 13: 507- 518.
- Konishi H, Kuroda S, Tanaka M, Matsuzaki H, Ono Y, Kameyama K, et al. Molecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem. Biophys. Res. Commun. 1995; 216: 526- 534.
- Mure H, Matsuzaki K, Kitazato KT, Mizobuchi Y, Kuwayama K, Kageji T, et al. Akt2 and AKT3 play a pivotal role in malignant gliomas. Neuro Oncol. 1979; 12: 221-232.
- Stein GH. T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979; 99: 43-54.
- Paul-Samojedny M, Suchanek R, Borkowska P, Pudelko A, Owczarek A, Kowalczyk M, et al. Knockdown of AKT3 (PKBγ) and PI3KCA suppresses cell viability and proliferation and induces the apoptosis of glioblastoma multiforme T98G cells. Biomed Res Int. 2014: 768181.
- Darzynkiewicz Z, Robinson JP, Crissman HA (eds.) Flow Cytometry. Methods in Cell Biology, Academic Press, Inc., San Diego. 1994.
- Henry S, George T, Hal B, Basiji D, Ortyn W, Morrissey P. Quantitative image based apoptotic index measurement using multispectral imaging flow cytometry: a comparison with standard photometric methods. Apoptosis. 2008; 13: 1054-1063.
- Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol. 2014; 31: 985.
- Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005; 19: 1751-1759.
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004; 64: 1079-1086.
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001; 20: 6969-6978.
- Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev. 2008; 34: 206-222.
- Chavez-Blanco A, Perez-Plasencia C, Perez-Cardenas E, Carrasco-Legleu C, Rangel-Lopez E, Segura-Pacheco B, et al. Antineoplastic effects of the DNA methylation inhibitor hydralazine and the histone deacetylase inhibitor valproic acid in cancer cell lines. Cancer Cell Int. 2006; 6: 2.
- Strey CW, Schamell L, Oppermann E, Haferkamp A, Bechstein WO, Blaheta RA. Valproate inhibits colon cancer growth through cell cycle modification in vivo and in vitro. Exp Ther Med. 2011; 2: 301-307.
- Xia Q, Sung J, Chowdhury W, Chen CL, Höti N, Shabbeer S, et al. Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo. Cancer Res. 2006; 66: 7237-7244.
- Das CM, Aguilera D, Vasquez H, Prasad P, Zhang M, Wolff JE, et al. Valproic acid induces p21 and topoisomerase-II (alpha/beta) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J Neurooncol. 2007; 85: 159-170.
- Li X-N, Shu Q, Su JM-F, Perlaky L, Blaney SM, Lau CC. Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol Cancer Ther. 2005; 4: 1912-1922.
- Kamitani H, Taniura S, Watanabe K, Sakamoto M, Watanabe T, Eling T. Histone acetylation may suppress human glioma cell proliferation when p21 WAF/Cip1 and gelsolin are induced. Neurooncology. 2002; 4: 95-101.
- Bacon CL, O’Driscoll E, Regan CM. Valproic acid suppresses G1 phasedependent sialylation of a 65kDa glycoprotein in the C6 glioma cell cycle. Int J Dev Neurosci. 1997; 15: 777-784.
- Knüpfer MM, Hernáiz-Driever P, Poppenborg H, Wolff JE, Cinatl J. Valproic acid inhibits proliferation and changes expression of CD44 and CD56 of malignant glioma cells in vitro. Anticancer Res. 1998; 18: 3585-3589.
- Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008; 269: 7-17.
- Hosein AN, Lim YC, Day B, Stringer B, Rose S, Head R, et al. The effect of valproic acid in combination with irradiation and temozolomide on primary human glioblastoma cells. J Neurooncol. 2015; 122: 263-271.
- Schwartz C, Palissot V, Aouali N, Wack S, Brons NH, Leners B, et al. Valproic acid induces non-apoptotic cell death mechanisms in multiple myeloma cell lines. Int J Oncol. 2007; 30: 573-582.
- Bollino D, Balan I, Aurelian L. Valproic acid induces neuronal cell death through a novel calpain-dependent necroptosis pathway. J Neurochem. 2015; 133: 174-186.
- Song K-S, Kim J-S, Yun E-J, Kim YR, Seo KS, Park JH, et al. Rottlerin induces autophagy and apoptotic cell death through a PKC-deltaindependent pathway in HT1080 human fibrosarcoma cells: the protective role of autophagy in apoptosis. Autophagy. 2008; 4: 650-658.
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011; 8: 528-539.
- Fu J, Shao C-J, Chen F-R, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neurooncology. 2010; 12: 328-340.
- Ouyang D-Y, Xu L-H, He X-H, Zhang YT, Zeng LH, Cai JY, et al. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy. 2013; 9: 20-32.
Citation: Paul-Samojedny M, Liduk E, Borkowska P, Kowalczyk M, Suchanek-Raif R, Zielinska A, et al. Valproic Acid (VPA) in Combination with Knockdown of AKT3 and PI3KCA Genes Inhibits Proliferation, Induces Apoptosis and Autophagy in T98G and U87MG Glioblastoma Multiforme Cells. Austin J Anat. 2021; 8(2): 1101.