
Research Article
Austin J Anesthesia and Analgesia. 2014;2(4): 1024.
The Evidence for Opioid-Induced Hyperalgesia Today
David A Edwards and Lucy Chen*
Department of Anesthesia, Critical Care & Pain Medicine, Harvard University, USA
*Corresponding author: Lucy Chen, Department of Anesthesia, Critical Care & Pain Medicine, Massachusetts General Hospital, Harvard University, Boston, MA, USA.
Received: March 14, 2014; Accepted: April 24, 2014; Published: April 28, 2014
Abstract
Hyperalgesia is an increased response to painful sensation. Opioid–induced hyperalgesia occurs after sustained opioid exposure and⁄or following abrupt cessation of opioid. At the cellular level, adenylyl cyclase superactivation and elevated cAMP levels lead to PKA–mediated enhanced neurotransmitter release. Spinal glutamate, substance P, and CGRP enhance pain through NMDA, NK–1, and CGRP receptors respectively. Non–opioid receptor mediated OIH may be caused by M3G activation of TLR4. In controlled human experiments OIH is evident to cold pain, but less consistently found with pain caused by other modalities such as heat, electricity, or pressure. Hyperalgesia detected in response to cold pain can be reduced by the NMDA antagonist ketamine. Patients on methadone maintenance also show an increased sensitivity to nociception between doses. Multi–modal analgesia may be the best strategy fortreatment of OIH in the clinical setting.
Keywords: Hyperalgesia; Opioid; Nociception; Ketamine
Introduction
Nearly all clinicians know that when they prescribe opioid medications to treat their patient’s pain, they are balancing against the risk of tolerance and addiction, overdose and death. What they know less about is that high–dose opioids, as an intra–operative infusion or as an oral home dose, could make the pain worse under certain conditions. This latter situation is called Opioid–Induced Hyperalgesia (OIH) and is defined as an increased response to a painful stimulus caused by exposure to opioids. The paradox of potentially doing harm when trying to provide relief is antithetical to a clinician’s sworn oath, so if real, must be recognized and avoided.
Is OIH real in humans, and if it is real, is it relevant? After 3 decades of research, the question is still being asked [1]. Since the early observation that patients on morphine had a lower threshold for pain [2,3], in vitro cell studies and in vivo animal studies have proceeded to uncover the potential mechanisms. Case reports and clinical studies of OIH are appearing with increased frequency. Models to empirically measure OIH in humans have been developed, and are used to detect hyperalgesia in these controlled situations. Still, what is its relevance in the clinic?
Clinically OIH may occur with chronic oral opioid consumption or after intra–operative intravenous infusions of opioid. In methadone aintained populations the threshold for pain sensation is decreased. After remifentanil infusions, post–operative hyperalgesia is presumed due to increased pain scores and opioid requirement. In either scenario, what are the clinician’s options? Withholding pain–relieving opioids is not the complete answer, but opioid–sparing techniques may be at least part of it.
It seems more obvious that the chronically treated patient may be subject to a relatively increased disservice by the prescription of opioids compared to the surgical patient that is given an intraoperative infusion. If methadone–maintained patients are more sensitive to pain, preventing ongoing suffering or managing perioperative pain becomes a challenge. Compare this to the patient exposed intra–operatively to a remifentanil infusion who is requiring more morphine in the recovery room. A transient increased use of morphine to become comfortable seems to be an appropriate measure and trade–off for preventing intra–operative severe surgical pain, especially if there are no long–term downsides. On the other hand, if there are options that can prevent hyperalgesia and also provide equal relief from pain then we are obligated to use them.
This review provides an overview of the evidence for OIH. First, in vitro cell models and in vivo animal models are reviewed followed by up–to–date evidence from human trials. Reference tables were created to show all human trials published to date. Several good reviews have been published on this topic that deal with the potential mechanisms in great depth [4,5]. This review emphasizes the evidence for OIH in humans in order to serve as a reference for clinicians.
Background
Hyperalgesia is an increased response to a painful stimulus. The injured organism is more sensitive to pain and thereby is encouraged to guard against further injury while the healing occurs. Chronic pain is the pathological extension of allodynia or hyperalgesia beyond the normal healing period.
The establishment of hyperalgesia is known to have a central mechanism. Peripheral injury may directly damage nerve and surrounding tissue inducing the release of an inflammatory milieu that facilitates transmission of pain. Continual pain stimulus results in plastic changes in the dorsal horn of the spinal cord that decrease the threshold to pain sensation for surrounding neural inputs. Descending pain–inhibiting pathways are themselves inhibited. Over time, as the wound heals and the barrage of peripheral pain stimuli decreases, hyperalgesia lessens and disappears. On the other hand, a continuous exogenous stimulus may cause hyperalgesia to persist. Continued delivery of exogenous opioids induces downstream mechanisms that result in opioid tolerance and hyperalgesia.
In vitro studies of the cellular mechanism of opioid–induced hyperalgesia
In vitro studies of OIH have used mammalian cell cultures or acute tissue preparations to reveal several potential underlying mechanisms (Table 1). Prolonged opioid exposure causes changes in opioid–receptor mediated, and opioid–receptor independent downstream second messenger systems that may persist beyond the duration of opioid exposure. Mechanisms underlying hyperalgesia may involve both primary neurons as well as changes in glia. We first look at the opioid receptor–mediated mechanisms.
![]()
Model
Representa.ve References
Chinese hamster ovary (CHO) cells
[9,13]
Dissociated dorsal root ganglion (DRG) of rat
[10,12,16-18,20,24]
African green monkey kidney COS77 cells
[15]
Premotor cardiac parasympatheLc nucleus ambiguous neurons of rat
[11]
Human embryonic kidney 293 (HEK7293) cells
[14,20,25]
Table 1: In vitro models used to study opioid/induced hyperalgesia.
µ–Opioid receptor mechanisms
Opioid receptors concentrate in peripheral nociceptive fibers that synapse centrally upon spinal dorsal horn neurons, and in pathways descending from the rostral ventral medial medulla to the spinal cord [6,7]. When exogenous opioids activate opioid receptors, a cascade of changes in down–stream second messenger systems leads to decreased neuronal transmission of pain (Figure 1A). Opioid receptors belong to the class of seven– transmembrane domain G–protein coupled inhibitory receptors, so when opioid ligand binds to the receptor they, in turn, activate Gi⁄o which dissociates into its α and β⁄γ subunits [8]. Gi⁄o α inhibits adenylyl cyclase (AC) activity, downstream levels of cyclic adenosine monophosphate (cAMP) are reduced, and protein kinase activity is reduced [9]. Potassium channels are made more permeable while calcium channels become less permeable, causing presynaptic terminals to hyperpolarize and decreasing the release of glutamate and substance P [7,10–12]. Postsynaptic terminals are also hyperpolarized resulting in decreased neuronal excitability and reduced signal transduction [7]. With respect to pain signal sensitivity and transmission, the neuronal cell becomes quiescent.
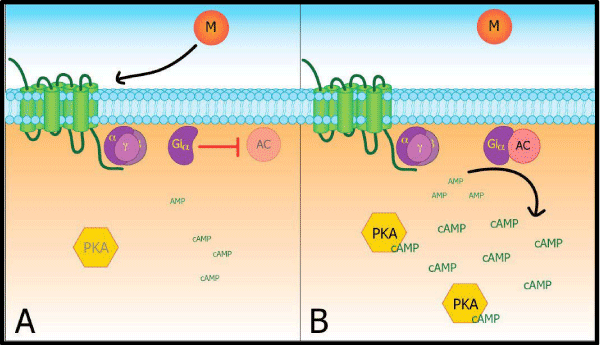
Figure 1: A. Morphine (M) activated the 7 transmembrane opioid receptor couple to Gi, which inhibits adenylyl cyclase (AC). B. In AC superactivation, even after morphine no longer acts on the opioid receptor, AC remains active cAMP levels are increased, and PKA is activated.
In chronic opioid exposure, in vitro studies show the situation is nearly reversed (Figure 1B). Chronic opioid activation of µ–, δ–, or ?– receptors transfected into neuronal cell types leads to Raf–1 mediated phosphorylation of AC [13]. Raf–1 phosphorylation of AC increases its activity [14], a process now commonly called AC “super activation” [15]. This leads to increased cAMP production and augmentation of protein kinase A (PKA), which in turn enhances the release of pain neurotransmitters such as calcitonin gene–related peptide [16–18]. Calcitonin gene–related peptide release may also be augmented by sustained morphine–induced AC sensitivity to excitatory pathways, such as prostaglandin E2 (PGE2) coupled to Gs [19].
Under short–term opioid exposure, the capsaicin and noxious heat sensitive transient receptor potential vanilloid 1 (TRPV1) voltage–gated ion channel is inhibited in the presence of low cAMP levels [20]. After sustained morphine exposure labeling of TRPV1 in the DRG is increased [21].
In this activated state, cellular signaling of pain is restored, and it now requires increased doses of opioid to reinstitute the quiescentstate. When opioids are stopped, either through washout or blocked, AC supersensitization persists and synaptic strength is facilitated in a process called opioid withdrawal long–term potentiation (LTP) [22,23]. Persistent AC supersensitization and opioid withdrawal LTP may underlie OIH.
Opioid receptor–independent mechanism
Opioid receptor independent mechanisms of OIH exist. Morphine–3–glucuronide (M3G), a liver metabolite of morphine, increases neuronal excitability via Toll–like receptors (TLR) [24,25]. Morphine activation of TLR4, concentrated in spinal microglia, causes the release of interleukin–1, and tumor necrosis factor (TNF–α) [25]. Toll–like receptor antagonists have been identified as possible treatments for alleviation of glial sustained hyperalgesia [26,27]. Suppression of inhibition by glia on lamina I neurons of the spinal dorsal horn may contribute to enhanced transmission and decreased threshold in these neurons during hyperalgesic states [28]. Ferrini showed prolonged exposure to morphine activated the purinoceptor, P2X4R, in microglia and caused the release of brainderived neurotrophic factor (BDNF) [28]. Block of BDNF receptor, tropomyosin related kinase B (TrkB), signaling prevented OIH and OIH was absent in mice lacking BDNF in microglia [28]. Morphine may also cause hyperalgesia independent of the µ–opioid receptor or M3G [29]. The complete story of morphine–induced hyperalgesia is not yet clear.
In vivo studies of opioid–induced hyperalgesia
Animal tests: In animals, experimental protocols to induce OIH consist of oral, subcutaneous (S.C), intravenous, intraperitoneal, or intrathecal injection of opioid [30]. Repetitive, continuous, or intermittent opioid dosing protocols can induce hyperalgesia after discontinuation or precipitated withdrawal by opioid receptor antagonists [31]. After prolonged high doses of opioid periods of hyperalgesia can last hours to days [30,32].
Hyperalgesia is measured by mechanical, thermal, or electrical nociceptive tests (Table 2). With OIH the withdrawal thresholds are reduced and withdrawal and response latencies are shorter for these tests. For a thorough list of animal studies of OIH see table 3 in Angst and Clark [30].
![]()
Modality
Model
Description
Representa.ve References
Mechanical pressure pain
Withdrawal threshold
Calibrated von Frey ?laments are applied to the hind foot and the smallest ?lament that elicits a withdrawal response is considered to be the withdrawal threshold.
[57]
Paw pressure withdrawal or vocalization tests
The threshold to paw withdrawal is measured while a pressure pad is applied to the metacarpal of the toe.
[63]
Thermal pain
Tail ?ick test
Radiant heat is applied to the tail or the tail is placed in hot water which causes the animal to ?ick its tail out of the way. The Lme to withdrawal of the tail is measured as the withdrawal latency.
[36,38,43,48]
Paw withdrawal test
Radiant heat is applied under the animal's foot. The time to withdrawal is measured. This test is more sensitive than the tail ?ick test.
[38]
Electrical pain
Hot plate test
An animal is placed on a hot plate of variable temperature. �When the animal licks its feet, or jumps o? of the plate the response latency is recorded.
[34,37]
Electrical shock tail vocalization test
The threshold to vocalization is measured with electrode7 delivered current to the tail.
[36]
Table 2: In vivo models used to study opioid/ induced hyperalgesia.
Peripheral delivery: In early animal investigations it was reported that repeated peripheral administration of morphine to guinea pigs caused hyperesthesia once the analgesia had worn off [33]. Kayan and Mitchell reported a decreased threshold to thermal hot plate test after repeated injection of morphine S.C. [34].
Central delivery: By injecting opiate intrathecally, the direct effects of centrally mediated analgesia and hyperalgesia are measured. While investigating the effects of spinal opioid pain relief, it was discovered that high doses of intrathecal morphine paradoxically caused hypersensitivity in rats [35–37]. The effect was reproduced by injection of morphine–3–glucuronide (M3G), indicating that both opioid receptor dependent and independent mechanisms of hyperalgesia exist [37].
Excitatory amino acid signaling in the spinal cord may be responsible for OIH just as it is for hyperalgesia and central sensitization due to peripheral nerve injury, chronic inflammation, and neuropathic pain [38–40]. Following 8 days of intrathecal morphine injection, rats developed tolerance and hyperalgesia measured by tail– flick and foot withdrawal tests. The hyperalgesia was prevented and reversed by intrathecal injection of dizocilpine, also known as MK 801, an N–methyl–D–aspartate (NMDA) receptor antagonist [38]. Single subcutaneous doses of heroin decreased pain threshold in rats which was blocked by NMDA antagonists MK 801 [41] and ketamine [31]. Fentanyl–induced hyperalgesia was also blocked by pretreatment with ketamine [32,42]. L–methadone caused thermal hyperalgesia, while the racemic isomer d–methadone prevented it, perhaps through its action on the NMDA receptor [43]. On the other hand, in one small study of methadone–induced thermal hyperalgesia memantine, a weaker NMDA antagonist than ketamine, failed to alter the nociceptive threshold [44]. Repeated morphine administration reduced spinal glutamate transporter, GLT–1, which is responsible for clearance of glutamate [45]. The beta–lactam antibiotic, ceftriaxone, inhibited OIH by up regulating GLT–1 [45]. NMDA receptor messenger RNA (mRNA) expression decreased during sub acute exposure to morphine, but returned to baseline with chronic treatment [46]. Ketamine co–administration prevented this effect on NMDA receptor mRNA expression [46]. Glycogen synthase kinase–3β (GSK–3β) modulates NMDA receptor trafficking. Inhibition of GSK–3β prevented remifentanil–induced hyperalgesia [47]. Enhanced glutamatergic signaling in the central nervous system is triggered by opioid exposure and measurable in behavioral tests of hyperalgesia, consistent with the aforementioned in vitro studies [36].
Excitability of nociceptive pathways in OIH extends beyond glutamatergic signaling systems. Neurokinin 1 (NK–1) cells are expressed in lamina I of the spinal dorsal horn and project to supraspinal areas that mediate pain processing [48]. Activated ascending NK–1 receptor expressing cells signal descending facilitation via serotonergic projections to the dorsal horn [49]. Opioid–induced hyperalgesia is characterized by an increase in spinal dynorphin [50], substance P [50,51] and calcitonin generelated peptide (CGRP) levels [49,51]. Substance P activates NK–1 receptors and causes internalization similar to what occurs in the spinal cord during chronic inflammation [51]. When injected intrathecally, substance P induced pain, while dynorphin antiserum prevented morphine–induced abnormal pain [50]. Ablation of NK–1 expressing cells prevented morphine–induced thermal hyperalgesia, FOS expression, and the up regulation of dynorphin [48]. Intrathecal injection of anti–inflammatory cyclooxygenase inhibitors blocked glutamate– and substance P–induced hyperalgesia [52]. Similarly intrathecal ibuprofen reversed hyperalgesia during opioid withdrawal [53]. Descending facilitation and OIH are blocked by serotonin antagonists, ondansetron and granisetron [48,49,51,54,55]. The excitatory state described in in vitro studies of OIH is supported by in vivo characterization of neuronal ascending and descending nociceptive signaling pathways.
While it is generally accepted now that OIH involves excitation of nociceptive pathways, there is still limited insight into the second messenger systems. Tumati extended their in vitro studies of the role of PKA through small interfering RNA (siRNA) knockdown of spinal PKA [56] in vivo. Knockdown attenuated sustained morphineinduced thermal hyperalgesia indicating that OIH is mediated by spinal PKA and cAMP mechanisms [56]. The downstream effects of opioid receptor activation also depend on protein kinase C (PKC) [40]. Intrathecal injection of the ganglioside GM1, a PKC inhibitor, along with morphine prevented OIH [38]. Fentanyl–induced hyperalgesia was prevented in knock–out mice lacking PKCγ gene [57]. Pretreatment of rats with intrathecal chelerythrine, a PKC inhibitor also prevented OIH [58].
The TRPV1 channel is inhibited by µ–opioid receptor mediated decreases in cAMP [20]. During opioid withdrawal, uncovered AC superactivation and elevated cAMP increased capsaicin–induced TRPV1 activity [20]. Subcutaneous morphine in TRPV1 wild–type mice produced thermal and tactile hypersensitivity but not in TRPV1 knockout mice [59]. This is consistent with in vitro studies mentioned above (Figure 1).
Opioid receptor independent mechanisms of OIH may exist. The morphine metabolite, M3G, is inactive at opioid receptors, yet is able to cause hyperalgesia [36,37,59]. M3G caused hyperalgesia without analgesia in opioid–receptor knockout mice [60]. Opioid-induced increases in spinal dynorphin may bind to bradykinin receptors and induce hyperalgesia. Sufentanil–induced hyperalgesia was found in wild–type but not bradykinin receptor (B2R) knockout mice [61].
Gabapentin and pregabalin act on the α2δ subunit of voltage gated calcium channels and are effective in the management of neuropathic pain, a modality involving central sensitization [62]. Intraperitoneal and intrathecal gabapentin dose dependently prevented a decrease in pain threshold in the paw pressure test after fentanyl–induced hyperalgesia [55,63,64]. Gabapentin acted synergistically to prevent OIH when combined with ketamine [42].
Genetic Susceptibility
Different rodent strains show differences in their susceptibility to OIH suggesting a genetic role. Spontaneously hypertensive rats (SHR) were more likely to develop hyperalgesia to hot plate test during morphine administration [65]. New genetic haplotype mapping techniques have identified variants of the beta– receptor and the P–glycoprotein transporter up–regulated in OIH that predispose certain animal strains to hyperalgesia [54,66,67]. Morphine inducedhyperalgesia was more pronounced, occurred earlier, and persisted longer in female, compared to male, Sprague–Dawley rats [68,69]. During morphine–induced hyperalgesia epigenetic regulation through histone acetylation seems to maintain and prolong the duration of hyperalgesia [70].
In summary, behavioral animal studies have provided insight into multiple mechanisms by which opioids may induce hyperalgesia. Centrally, the mechanism is NMDA dependent. Opioid–induced hyperalgesia is characterized by enhancement of nociceptive signaling via ascending and descending pathways in the spinal cord. Animal studies agree with cell models, that there are both PKA and PKC contributions to OIH. In vivo studies introduced the possibility that genetic variance plays a role in mammalian susceptibility to OIH.
Human studies of opioid–induced hyperalgesia
Clinically, hyperalgesia has been recognized for over a century [2,3]. With the introduction of several newer opioid agonists into clinical practice, a different propensity toward hyperalgesia has been noticed. Most of the early observational studies, case reports, and clinical trials of hyperalgesia involved patients on methadone or remifentanil (Table 3), opioid agonists of very different pharmacokinetic and dynamic properties. Remifentanil is an ultrashort acting opioid analgesic commonly used as an intra–operative infusion. Remifentanil–induced opioid hyperalgesia occurs after the infusion has been stopped in the recovery room and thereafter. Methadone, on the other hand, is prescribed as a maintenance medication in lieu of opioids of abuse. Early observational research showed that patients on chronic methadone may be more sensitive to pain [71–73]. Clinical studies of OIH look at its expression under these 2 relative conditions: in the immediate post–surgical phase following intra–operative infusions of opioids, or during chronic oral consumption. Our search found 21 clinical studies of remifentanilinduced hyperalgesia (Table 3), 10 studies of hyperalgesia in patients on methadone, and 10 remaining studies of other opioids (Table 3), not including case reports.
![]()
Reference
Model
Study Design
Opioid
Route
Outcome Measure
Details, Results
[85]
remifentanil infusion for major abdominal surgery
randomized, �controlled,
remifentanil
IV
postop VAS and morphine consumption
n=50, high intraop remifentanil dose caused increased postop VAS and morphine �consumption
[74]
remifentanil infusion; heat and capsaicin-induced hyperalgesia (punctate hyperalgesia with von Frey), thermode induced heat pain
Double-blinded, placebo-controlled
remifentanil
IV
VAS, area of punctate hyperalgesia
n=14, post-remifentanil infusion VAS was increased to heat pain, area of hyperalgesia was increased compared to placebo, but not compared to pre-remi baseline
[75]
remifentanil infusion vs. remi+ketamine; IM needle stim, transcutaneous electrical stim, pressure pain
randomized, double-blind, placebo-controlled, crossover
remifentanil
IV
pain threshold, pain tolerance
n=12, post-remifentanil infusion patients have lower tolerance to pressure pain not prevented with ketamine, none or increased tolerance to electrical stim
[78]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey)
randomized, double-blind, placebo-controlled
remifentanil
IV
area of punctate hyperalgesia
n=13, male, post-remifentanil infusion area of punctate hyperalgesia expanded, but not post ketamine or clonidine infusion
[77]
remifentanil infusion; heat and capsaicin� induced hyperalgesia (punctate hyperalgesia with von Frey)
remifentanil
IV
area of punctate hyperalgesia
n=10, post-remifentanil infusion area of punctate hyperalgesia expanded at 4 hours
[81]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey), transdermal electrode stim
randomized, double-blind, placebo-controlled, cross over
remifentanil
IV
NRS, area of punctate hyperalgesia
n=13, post-saline control slightly expanded area of hyperalgesia but not NRS, whereas post-remifentanil infusion area of hyperalgesia greatly expanded but not NRS; post-naloxone infusion expanded area of hyperalgesia but by less
[79]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with probe), thermode induced heat pain
randomized, double-blind, placebo-controlled, �cross-over
remifentanil
IV
VAS, area of punctate hyperalgesia
n=X, post remifentanil infusion area of punctate hyperalgesia expanded, but no increase in VAS with heat pain; post-ketamine+remi infusion abolished expansion of hyperalgesia
[68]
major abdominal surgery randomized to remifentanil at low dose, large dose, or large dose + ketamine; mechanical pain threshold to von Frey ?lament, pressure pain threshold
randomized, �controlled
remifentanil
IV
VAS, NRS, pain threshold, 48- hr morphine consumption, time-to-?rst morphine
n=75 (25 patients per group), post large dose remi infusion tactile pain threshold was less at 24 and 48 hrs. No di? in pressure pain thresholds. No di? in VAS and NRS. No di? in time to ?rst morphine needed in PACU nor total PACU consumption, but 48-hr consumption was sig. greater in large dose remi.
[86]
major abdominal surgery randomized to remifentanil infusion or placebo while under general plus epidural anesthesia
randomized, double-blind, placebo-controlled
remifentanil
IV
VAS, 24-hr morphine consumption
n=50, post remifentanil infusion patients had higher VAS, but no di?erence in 24 hour PCA morphine consumption
[105]
pediatric patients undergoing surgical correction of idiopathic scoliosis under remifentanil infusion or IV morphine
prospective, �randomized, double-blind
remifentanil, morphine
IV
NRS, 24-hr morphine consumption
n=30 (15 remi, 15 morphine), 24-hr morphine consumption was sig. more in the remifentanil group. There were no di?erences in NRS.
[80]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey)
randomized, double-blind, placebo-controlled, �cross-over
remifentanil
IV
NRS, area of punctate hyperalgesia
n=15, male, post remifentanil infusion area of punctate hyperalgesia expanded and NRS pain intensity increased; pre-remi infusion parecoxib diminished remi- induced area of hyperalgesia, but parallel administered parecoxib did not
[82]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey)
randomized, double-blind, placebo-controlled, �cross-over
remifentanil
IV
NRS, area of punctate hyperalgesia
n=15, post remi area of punctate hyperalgesia expanded and NRS increased; post-propofol infusion did not expand area of hyperalgesia or cause increased NRS; propofol co-admin weakened but did not prevent post-remi hyperalgesia
[104]
pediatric patients for surgical correction of idiopathic scoliosis under remi infusion +/ pre-remi injection of IV morphine
randomized, double-blind, placebo-controlled
remifentanil
IV
NRS, 24-hr morphine consumption
n=40 (18 in morphine+remi, 19 in remi group) no difference in morphine consumption or NRS scores between groups
[19]
breast cancer surgical patients under high vs. low remifentanil infusion with either sevo?urane or propofol
randomized, �blinded, controlled
remifentanil
IV
VAS, 24-hr morphine consumption
n=214 (female, 50 in each group), morphine consumption was higher in sevo
+high remi group compared to sevo+low remi, and both propofol groups. VAS was also higher in the sevo+high remi group vs. all other groups
[83]
remifentanil infusion; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey), CPT
randomized, double-blind, placebo-controlled, �cross-over
remifentanil
IV
NRS, area of punctate hyperalgesia
n=16, male, post remi area of punctate hyperalgesia expanded and NRS increased; pretreatment with parecoxib or ketorolac decreased area of post-remi hyperalgesia, e?ect of parecoxib was greater than ketorolac; NRS after remi infusion was increased and not prevented by parecoxib or ketorolac
[87]
Robot-assisted laparoscopic prostatectomy under remifentanil infusion; remi+magnesium injection to incision site vs. remi+saline injection vs. no remi+saline injection
randomized, placebo- controlled
remifentanil
IV
VAS,� time-to-post-op requirement of opioid, total post-op opioid requirement
n=75 (25 remi+mag, 25 remi+saline, 25 des+saline), time to ?rst analgesic was longer in the mag. and des. groups, VAS was lower in mag. and des. groups for 24 hrs, total opioid consumption was lower in mag. and des groups.
[99]
Laparoscopic gynecological surgery under sevo?urane and remifentanil infusion with or without ketamine
cohort, controlled (not controlled to evaluate remifentanil)
remifentanil
IV
VAS, post-op morphine requirement
n=40 (female. 20 remi+ketamine, 20 remi only), VAS was lower in the ketamine treated group up to 3 hours post op, PCA morphine consumption was reduced in the ketamine group up to 2 hours post-op
[42]
elective open septorhinoplasty surgery under propofol general anesthesia with remifentanil infusion +/ N2O; mechanical pain threshold with von Frey ?lament
randomized, double-blind, controlled (not controlled to evaluate �remifentanil)
remifentanil
IV
VAS, post-op morphine requirement, mechanical pain threshold
n=50 (25 with N2O, 25 with O2�as control), post remifentanil no di?erence in VAS or morphine consumption with or without N2O intraop. �N2O group had lower post op mechanical pain threshold at 12-18 hours but not at 2 hours
[103]
remifentanil infusion; thermode induced heat pain, mechanical hyperalgesia with punctate probes
crossover, placebo-controlled, blinded
remifentanil
IV
VAS
n=33 (10 excluded), no sig. di?erence in VAS to heat or punctate pain
[84]
remifentanil infusion in patients +/- propranolol; transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey), heat pain
randomized, double-blind, placebo-controlled, crossover
remifentanil
IV
area of punctate hyperalgesia , pain threshold, pain tolerance
n=10, post remifentanil infusion area of punctate, but not thermal, hyperalgesia greatly expanded. Punctate hyperalgesia was prevented post remi+propranolol infusion.
[87]
intraop remifentanil with dexmedetomidine or placebo in laparoscopic assisted vaginal hysterectomy patients; mechanical hyperalgesia with von Frey
randomized, placebo-controlled
remifentanil
IV
pain threshold, pain tolerance, VAS, morphine consumption
n=90 (30 placebo+remi, 30 placebo+high remi, 30+dex+high remi), post-remi infusion pain threshold was lower in placebo+high remi group, VAS and morphine requirement were also higher post op in placebo+high remi group; dex group showed no hyperalgesia
Table 3: Clinical Research Trials of Opioid-Induced Hyperalgesia with Remifentanil.
Hyperalgesia during remifentanil withdrawal
Hyperalgesia in the period after remifentanil infusion is the most frequently tested model for studying OIH in humans. Unfortunately most studies are underpowered, testing methods differ, and outcomes are inconsistent. In a series of small, randomized, controlled trials volunteers underwent remifentanil infusion under experimental conditions [74–80]. In two studies remifentanil hyperalgesia was evaluated by testing the expansion of a localized area of hyperalgesia created with heat and capsaicin [74,77]. Post–infusion painscores measured by visual analog scale (VAS) were increased to heat pain using a thermode [75] and the measured area of hyperalgesia was increased up to 4 hours after infusion [74,77]. Area of hyperalgesia was also expanded after remifentanil infusion in a model using transcutaneous electrical stimulation to create a region of hyperalgesia [75,78–84]. In 2 of these studies pain intensity scores were not significantly different from controls [79,81] but were significant in 3 studies [80,82,83].
Prospective studies of surgical patients where remifentanil was used have enrolled more patients and consistently shown increased post–operative pain [85–87]. Only Guignard et al. [85] and Lee et al. [87] found an increase in post–operative morphine consumption.
Hyperalgesia in patients on methadone
Prospective trials analyzed pain tolerance in opioid addicted patients transitioned to methadone maintenance, or patients with chronic pain on methadone (Table 4). In most trials methadone users were less tolerant of cold pain during the cold pressor test than nonusers [71,72,88–93] but in Pud et al. threshold to cold pain was increased [94]. No hyperalgesia to mechanical pain tests was found in patients on methadone [92,95]. Two underpowered studies found methadone maintained patients to be less tolerant but have unchanged pain detection thresholds to electrical stimuli [72,95], while another showed increased threshold to electrical pain 92]. Methadone–induced hyperalgesia is poorly supported. Most of the evidence for it comes from too few investigating groups with conflicting outcomes.
![]()
Reference
Model
Study Design
Opioid
Route
Outcome Measure
Details, Results
[71]
methadone maintained heroin dependent; CPT
methadone
PO
pain threshold
pain threshold lower in drug free ex-addicts and methadone maintained patients compared to controls
[73]
heroin users on methadone maintenance; mechanical pressure test
cohort, match controlled
I-methadone
PO
pain threshold
n=42, pain threshold and tolerance similar to controls at time of plasma trough
[89]
Methadone-maintained opioid abusers response to hydromorphone; CPT
cohort, placebo-controlled, 2-way factorial, mixed model
methadone maintained, hydromorphone tested
PO
pain tolerance before and after hydromorphone
n=120 (60 methadone, 60 controls), methadone maintained patients less tolerant than controls, no sig. di?erence with hydromorphone analgesia vs. NSAID
[95]
Methadone-maintained patients; CES, CPT
cohort, match controlled
methadone
PO
pain threshold, pain tolerance
n=32 (16 methadone, 16 controls), methadone-maintained are less tolerant to electrical stim and cold pain, but have lower threshold to cold pain only
[72]
Methadone-maintained heroin users; CES, CPT
cohort
methadone maintained, morphine tested
IV
pain threshold, pain tolerance
n=8 (4 methadone/morphine, 4 controls), methadone patients have lower threshold and tolerance to CPT pain, no di?erence in threshold but lower tolerance for CES pain
[90]
Opioid-maintained former opiate addicts, CPT
cohort
methadone, buprenorphine
PO
pain tolerance
n=54 (18 methadone, 18 buprenorphine, 18 control), methadone and buprenorphine maintained patients are equally less tolerant
[60]
opioid dependent patients on methadone maintenance on once daily doses (11-45mg, 46-80mg, 81-115mg) compared to controls; CPT, CES
Double-blind, placebo controlled, crossover
methadone
PO
pain threshold, pain tolerance
n=28 (18 methadone maintained, 10 control), pain threshold and tolerance increased in controls with morphine, but did not on methadone maintained patients
[94]
heroin or methadone addicted patients compared to healthy non-addicts, CPT
cohort, controlled
heroin, methadone
IV, PO
VAS, pain threshold, pain tolerance
n= �(60 opioid addicts, 70 controls), opioid addicts had lower VAS,
increased threshold but decreased tolerance
[98]
methadone maintained patients treated with dextromethophan or placebo; CPT, CES
Double-blind, placebo-controlled
methadone
PO
pain threshold, pain tolerance
n=40 (18 dex, 22 placebo), no di?erence in pain threshold or tolerance between dex patients and placebo
[44]
methadone maintained patients for dependence compared to patients with chronic pain on methadone or morphine; mechanical pain threshold with von Frey ?lament, CPT, CES
Observational
methadone, morphine
PO
pain threshold, pain tolerance
n=?, no di?erence between treated groups, but all treated groups (methadone or morphine) showed lower threshold and decreased tolerance to cold pain. There was no hyperalgesia to electrical pain.
[101]
methadone maintained individuals treated with gabapentin vs placebo; CPT
randomized, placebo-controlled
methadone
PO
pain threshold, pain tolerance
n=26 (10 gabapentin, 16 placebo), patients on gabapentin had increased threshold and increased tolerance to cold pain
[91]
heroin addicts randomized to methadone or buprenorphine treatment with matched drug-free controls; pain measured at trough and peak opioid plasma levels using CPT, electrical stimulation
randomized, match-controlled
methadone, buprenorphine
PO
pain threshold, pain tolerance
n=82 (11 methadone, 64 buprenorphine, 21 controls), baseline threshold and tolerance to cold were less in heroin-dependent group, hyperalgesia increased at trough in both methadone and buprenorphine groups.
[92]
Opioid-dependent subjects (methadone or buprenorphine); CPT, CES, mechanical pressure test, ischemic pain test
open design, cohort, controlled
methadone, buprenorphine
PO
pain threshold, pain tolerance
n=32 (8 on methadone, 8 on buprenorphine, 16 matched controls), pain threshold and tolerance were reduced in both methadone and buprenorphine groups; electrical pain thresholds increased in opioid groups but with no change in tolerance; no di?erences in ischemic or mechanical pain tests
[93]
male, heroin or methadone addicts, former addicts, drug-naïve; CPT
Observational
heroin, methadone
PO
pain threshold, pain tolerance
n=143 (50 heroin or methadone, 43 former addicts, 50 controls), active opioid users show decreased pain threshold and tolerance while former addicts do not
Table 4: Clinical Research Trials of Opioid-Induced Hyperalgesia with Methadone.
Hyperalgesia reports with other opioids
Of the remaining published studies of OIH, 2 are prospective, randomized, double–blinded, and placebo–controlled trials (Table 5) [75,96]. The first trial was done in a crossover format involving 12 patients exposed to an alfentanil infusion [76]. The area of focal hyperalgesia induced by transcutaneous electrical stimulation was measured before and after alfentanil infusion and found to be insignificant. Chu et al. performed a preliminary observational trial of patients with chronic low back pain and showed that after 1– month of oral morphine, pain threshold and tolerance to cold, but not heat, pain were reduced [97]. The follow–up controlled trial randomized 102 chronic low back pain patients to 1– month of morphine treatment versus placebo, and then evaluated hyperalgesia after remifentanil infusion [96]. No difference was found between groups for cold or heat pain. On the other hand, one study found that decreased heatpain threshold and exacerbated temporal summation of the second pain may be characteristic quantitative sensory test (QST) changes in subjects with chronic opioid therapy [40].
![]()
Reference
Model
Study Design
Opioid
Route
Outcome Measure
Details, Results
[88]
opiate vs. cocaine abusers; CPT
Quasi-experimental
opioid
PO
pain tolerance
n=122, male, pain intolerance correlated with opioid use
[31]
intraop high and low dose fentanyl infusion for TAH
fentanyl
IV
postop VAS and fentanyl consumption
n=60, female, higher intraop fentanyl caused increased postop VAS and fentanyl consumption
[76]
alfentanil infusion, transcutaneous electrical stim-induced hyperalgesia (punctate hyperalgesia with von Frey)
randomized, double-blind, placebo-controlled, crossover
alfentanil
IV
NRS, area of punctate hyperalgesia
n=12, post-alfentanil infusion patients showed a trend (insig) toward increased area of hyperalgesia but baseline NRS, whereas post-ketamine (sig) or lidocaine (insig) infusion caused reduced area of hyperalgesia
[90]
ADP protocol with IM morphine, IV morphine, hydromorphone, CPT
Quasi-experimental placebo-controlled crossover
morphine, hydromorphone
IM, IV
pain threshold, pain tolerance
n=4, male, IM and IV morphine and IV hydromorphone lowered pain threshold and tolerance following naloxone-induced withdrawal (ADP protocol)
� [100]
chronic non-malignant pain or cancer-related pain patients on opioids for 3+ months vs. NSAIDS; mechanical pain threshold with von Frey ?lament, mechanical pain threshold, heat pain threshold, suprathreshold heat pain NPS
cohort, tester blind, controlled
opioid
PO
NPS, pain threshold
n=224 (142 on opioid, 82 non-opioid), no di?erence in pain threshold, no di?erence in NPS in suprathreshold heat pain
[97]
chronic low back pain patients on oral morphine; CPT, heat pain
prospective, observational
morphine
PO
pain threshold, pain tolerance
n=6, 1-month oral morphine lowered pain threshold and tolerance to CPT, but not to heat
[25]
chronic non-malignant pain or cancer-related pain patients on chronic opioids compared to patients with chronic nonmalignant pain on chronic non- opioid analgesia; CPT
cohort
opioid (oxycodone, morphine, tramadol, propoxyphene, fentanyl, codeine)
PO
NPS, pain threshold, pain tolerance
n=110 (73 opioid treated group, 37 non-opioid treated control), chronic opioid treated group showed no decreased threshold, no di?erence in pain tolerance, and no di?erence in pain intensity.
[101]
chronic pain patients on opioids undergoing outpatient opioid tapering; heat pain
prospective, cohort
opioid (reported morphine equiv. dose)
PO
NRS
n=109, baseline opioid using patients showed decreased threshold to pain, tapering of opioid resulted in decreased heat pain compared to baseline
[84]
chronic low back pain tested post-remifentanil infusion, then treated for 1 month morphine vs. placebo and retested post-remi infusion; CPT, heat pain
randomized, double-blind, placebo-controlled
Sustained-release morphine
PO
pain threshold, pain tolerance
n=102, post-remifentanil infusion pain threshold and tolerance were not di?erent after 1 month of oral morphine
[87]
cancer patients on opioids vs. not on opioids undergoing interventional pain procedure tested with pain sensation to lidocaine injection
Observational
opioid (oxycodone, morphine, hydromorphone, codeine, fentanyl)
PO, TD
BPI, NRS,
unpleasantness score, behavior pain score
n=82 (62 opioid, 20 non-opioid), lidocaine injection NRS, unpleasantness, and behavior pain score were increased in opioid group, negative correlation between functional status and post-injection NRS
[61]
patients with moderate to severe chronic lumbar radicular pain, compared before and after 4 weeks of opioid therapy; CPT, phasic heat pain
prospective, open- label preliminary study
hydromorphone
PO
VAS, pain threshold, pain tolerance
n=40 (30 on hydromorphone, 10 controls),patients on hydromorphone had increased VAS to heat pain,
Table 5: Clinical Research Trials of Opioid-Induced Hyperalgesia with Other Opioids.
Translational trials
Cellular and animal models of OIH suggest that chronic opioid exposure can result in paradoxical neuronal excitation mediated by the NMDA receptor and many second messenger systems. Ketamine and dextromethorphan are NMDA antagonists studied in the context of OIH in human trails [75,76,78,79,98,99]. In a randomized crossover trial Koppert et al reported decreased area of punctate hyperalgesia after ketamine infusion [76], supported by 2 subsequent trials [78,79]. In another randomized crossover study Luginbuhl et al reported a lower tolerance to pressure pain after remifentanil infusion that was ot prevented by ketamine [75]. In methadone maintained patients there was no difference in pain threshold or tolerance to cold in those taking dextromethorphan versus placebo [98].
Cyclooxygenase inhibitors may reduce hyperalgesia via reduction in PGE2 activity on the NMDA receptor of astrocytes and spinal cord dorsal horn neurons [80]. In a randomized controlled trial, parecoxib given before remifentanil infusion, but not during, reduced the area of punctate hyperalgesia [80]. Parecoxib was more effective that ketorolac in reducing the area of hyperalgesia after remifentanil [83]. Reznikov compared the treatment of 224 patients with chronic pain over 3 months using opioids or non–steroidal anti– inflammatory medications and found no difference in pain intensity to mechanical or heat pain [100].
Discussion
Although originally OIH was detected by astute clinicians observing hyperesthesia in their patients, controlled human studies are far from clear and the evidence for OIH is strongest and most consistent in cellular and animal models. The phenomenon of persistent AC superactivation by opioids, and subsequent neuronal excitation may be the underlying mechanism of OIH, as far as it has been shown to occur in rodents. Discovery that NMDA antagonists prevent OIH has led to human trials using ketamine to antagonize remifentanil– induced hyperalgesia. This is a practical first step as both medications are commonly used intra–operatively; however, the dissociative side effects of ketamine limit its use in other realms. Gabapentin and pregabalin are also used frequently for the treatment of chronic pain and occasionally in the perioperative setting. Their action at α2δ receptors to inhibit calcium channels decreases hyperalgesia in rodents [42,55,64] and potentially in humans [101]. Adrenergic antagonists clonidine, dexmedetomidine, and propranolol may have promise against hyperalgesia both in the operating room and the clinic [78,84,87]. Medications that target second messenger systems involved in OIH have yet to be employed in clinical trials.
The relevance of mild transient hyperalgesia that may occur after remifentanil infusions in the operation room has been questioned [1,102]. In practice, remifentanil is rarely used alone, and due to its short duration of action most practitioners add a long–acting opioid to treat pain in the recovery room. In neurosurgical and orthopedic surgeries that require motor and sensory evoked potential monitoring, remifentanil and propofol combination is common. Propofol too, may reduce hyperalgesia caused by remifentanil [84]. Quantitative sensory tests to detect hyperalgesia in controlled experiments may not be comparable to the perioperative setting. After remifentanil infusions most studies show hyperalgesia to cold but questionably to heat, electrical, and pressure pain [75,79,96,103]. Postoperatively, hyperalgesia has been presumed given an increase in 24–hour opioid consumption despite conflicting or insignificant pain intensity scores [87,104,105].
Hyperalgesia may be more relevant in the chronic pain setting. If prolonged opioid abuse or maintenance results in hypersensitivity to nociception, any offense such as injury or surgery can result in a relative increase in suffering compared to the non–opioid user. Moreover, rapidly increasing opioids, without concomitant addition of adjuvants that prevent hyperalgesia, may expose a patient to increased pain in the presence of a continuous nociceptive stimulus. Rapidly escalating the dose of opioid not only exposes a patient to greater risks but also hastens tolerance, and can lead to rapid onset of hyperalgesia. Opioid sparing techniques such as opioid rotation may allow for similar or improved pain control without inducing neuroexcitation as seen with rapid opioid escalation [106,107,108].
In humans, OIH is still a diagnosis of exclusion. There is no empiric test used clinically to diagnose a patient with OIH. Clinicians may diagnose patients with OIH based upon opioid use history, response to opioids, physiological signs of hyperalgesia, a patient’s subjective complaints, and exclusion of other possible causes of decreased threshold to pain. Although OIH is thought of more often in patients on chronic opioids, it may also occur in the acute setting as well [78]. Patients may experience an increase in pain with increase in opioid as well as a decreased threshold for detection of pain, two signs hat differentiate tolerance from hyperalgesia. Objective physiological signs may be evident; a patient’s heart rate, respiratory rate, and level of awareness may paradoxically increase despite escalating doses of opioid, and without disease progression. A prudent strategy to avoid OIH would be to employ multi modal analgesia in both the operating room and the clinic, avoid rapid dose escalation of opioids, and instead employ the principle of opioid rotation for patients who continue to suffer despite high and escalating dose of opioid.
References
- Martinez V, Fletcher D. Prevention of opioid-induced hyperalgesia in surgical patients: does it really matter? Br J Anaesth. 2012; 109: 302-304.
- Landsberg E. Investigations into the fate of morphine in the living organism. P?üger, Archive for the Total Physiology of man and animals. 1880; 23: 413-433.
- Andrews HL. The effect of opiates on the pain threshold in post-addicts. J Clin Invest. 1943; 22: 511-516.
- Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain002E 2008; 24: 479-496.
- Mao J. Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep. 2006; 10: 67-70.
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, Christie MJ. Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J Neurosci. 2002; 22: 10847-10855.
- Yaksh TL, Noueihed R. The physiology and pharmacology of spinal opiates. Annu Rev Pharmacol Toxicol. 1985; 25: 433-462.
- Birnbaumer L, Abramowitz J, Brown AM. Receptor-effector coupling by G proteins. Biochim Biophys Acta. 1990; 1031: 163-224.
- Avidor-Reiss T, Bayewitch M, Levy R, Matus-Leibovitch N, Nevo I, Vogel Z. Adenylyl cyclase super sensitization in mu-opioid receptor-transfected Chinese hamster ovary cells following chronic opioid treatment. J Biol Chem. 1995; 270: 29732-29738.
- Akins PT, McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993; 56: 759-769.
- Irnaten M, Aicher SA, Wang J, Venkatesan P, Evans C, Baxi S, et al. Mu- opioid receptors are located postsynaptically and endomorphin-1 inhibits voltage-gated calcium currents in premotor cardiac parasympathetic neurons in the rat nucleus ambiguus. Neuroscience, 2003; 116: 573-582.
- Schroeder JE, McCleskey EW. Inhibition of Ca2+ currents by a mu-opioid in a de?ned subset of rat sensory neurons. J Neurosci. 1993; 13: 867-873.
- Varga EV, Rubenzik MK, Stropova D, Sugiyama M, Grife V, Hruby VJ, et al. Converging protein kinase pathways mediate adenylyl cyclase superactivation upon chronic delta-opioid agonist treatment. J Pharmacol Exp Ther. 2003; 306: 109-115.
- Tan CM, Kelvin DJ, Litch?eld DW, Ferguson SS, Feldman RD. Tyrosine kinase-mediated serine phosphorylation of adenylyl cyclase. Biochemistry. 2001; 40: 1702-1709.
- Avidor-Reiss T, Nevo I, Levy R, Pfeuffer T, Vogel Z. Chronic opioid treatment induces adenylyl cyclase V superactivation. Involvement of Gbetagamma. J Biol Chem. 1996; 271: 21309-21315.
- Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced calcitonin gene-related peptide and substance P increases in cultured dorsal root ganglion neurons. Neuroscience. 2000; 99: 529-539.
- Yue X, Tumati S, Navratilova E, Strop D, St John PA, Vanderah TW, et al. Sustained morphine treatment augments basal CGRP release from cultured primary sensory neurons in a Raf-1 dependent manner. Eur J Pharmacol. 2008; 584: 272-277.
- Tumati S, Yamamura HI, Vanderah TW, Roeske WR, Varga EV. Sustained morphine treatment augments capsaicin-evoked calcitonin gene- related peptide release from primary sensory neurons in a protein kinase A- and Raf-1- dependent manner. Journal of Pharmacology and Experimental Therapeutics. 2009; 330: 810- 817.
- Tumati S, Roeske WR, Vanderah TW, Varga EV. Sustained morphine treatment augments prostaglandin E2-evoked calcitonin gene-related peptide release from primary sensory neurons in a PKA-dependent manner. European Journal of Pharmacology. 2010; 648: 95-101.
- Spahn V, Fischer O, Endres-Becker J, Schäfer M, Stein C, Zöllner C. Opioid withdrawal increases transient receptor potential vanilloid 1 activity in a protein kinase A-dependent manner. Pain. 2013; 154: 598-608.
- Vardanyan A, Wang R, Vanderah TW, Ossipov MH, Lai J, Porreca F, et al. TRPV1 receptor in expression of opioid-induced hyperalgesia. J Pain. 2009; 10: 243-252.
- Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci U S A. 1975; 72: 3092-3096.
- Heinl C, Drdla-Schutting R, Xanthos DN, Sandkühler J. Distinct mechanisms underlying pronociceptive effects of opioids. J Neurosci. 2011; 31: 16748- 16756.
- Due MR, Piekarz AD, Wilson N, Feldman P, Ripsch MS, Chavez S, et al. Neuroexcitatory effects of morphine-3-glucuronide are dependent on Toll- like receptor 4 signaling. J Neuroin?ammation. 2012; 9: 200.
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, et al. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience. 2010; 165: 569-583.
- Li Q. Antagonists of toll like receptor 4 maybe a new strategy to counteract opioid-induced hyperalgesia and opioid tolerance. Med Hypotheses. 2012; 79: 754-756.
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. Scienti?cWorldJournal. 2007; 7: 98-111.
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, Castonguay A. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl homeostasis. Nat Neurosci. 2013; 16: 183-192.
- Swartjes M, Mooren RA, Waxman AR, Arout C, van de Wetering K, den Hartigh J, et al. Morphine induces hyperalgesia without involvement of μ-opioid receptor or morphine-3-glucuronide. Mol Med. 2012; 18: 1320- 1326.
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006; 104: 570-587.
- Laulin JP, Célèrier E, Larcher A, Le Moal M, Simonnet G. Opiate tolerance to daily heroin administration: an apparent phenomenon associated with enhanced pain sensitivity. Neuroscience. 1999; 89: 631-636.
- Célèrier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, et al. Long- lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiology. 2000; 92: 465-472.
- Mulé SJ, Clements TH, Layson RC, Haertzen CA. Analgesia in guinea pigs: a measure of tolerance development. Arch Int Pharmacodyn Ther. 1968; 173: 201-212.
- Kayan S, Woods LA, Mitchell CL. Morphine-induced hyperalgesia in rats tested on the hot plate. J Pharmacol Exp Ther. 1971; 177: 509-513.
- Tang AH, Schoenfeld MJ. Comparison of subcutaneous and spinal subarachnoid injections of morphine and naloxone on analgesic tests in the rat. Eur J Pharmacol. 1978; 52: 215-223.
- Woolf CJ. Intrathecal high dose morphine produces hyperalgesia in the rat. Brain Res. 1981; 209: 491-495.
- Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: clinical and theoretic implications. Anesthesiology. 1986; 64: 590-597.
- Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994; 14: 2301-2312.
- Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991; 44: 293-299.
- Chen L, Huang LY. Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a mu opioid. Neuron. 1991; 7: 319-326.
- Laulin JP, Larcher A, Célèrier E, Le Moal M, Simonnet G. Long-lasting increased pain sensitivity in rat following exposure to heroin for the ?rst time. Eur J Neurosci. 1998; 10: 782-785.
- Van Elstraete AC, Sitbon P, Benhamou D, Mazoit JX. The median effective dose of ketamine and gabapentin in opioid-induced hyperalgesia in rats: an isobolographic analysis of their interaction. Anesth Analg. 2011; 113: 634- 640.
- Holtman JR Jr, Wala EP. Characterization of the antinociceptive and pronociceptive effects of methadone in rats. Anesthesiology. 2007; 106: 563-571.
- Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. 2009; 10: 316-322.
- Chen Z, He Y, Wang ZJ. The beta-lactam antibiotic, ceftriaxone, inhibits the development of opioid-induced hyperalgesia in mice. Neurosci Lett. 2012; 509: 69-71.
- Ohnesorge H, Feng Z, Zitta K, Steinfath M, Albrecht M, Bein B. In?uence of clonidine and ketamine on m-RNA expression in a model of opioid-induced hyperalgesia in mice. PLoS One. 2013; 8: e79567.
- Li Y, Wang H, Xie K, Wang C, Yang Z, Yu Y, et al. Inhibition of glycogen synthase kinase-3β prevents remifentanil-induced hyperalgesia via regulating the expression and function of spinal N-methyl-D-aspartate receptors in vivo and vitro. PLoS One. 2013; 8: e77790.
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007; 129: 35-45.
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Super?cial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002; 5: 1319-1326.
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP Jr, et al. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent ?bers. J Neurosci: the of?cial journal of the Society for Neuroscience. 2002; 22: 6747-6755.
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP Jr, et al. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005; 116: 276-288.
- Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992; 257: 1276-1279.
- Dunbar SA, Karamov IG, Buerkle H. The effect of spinal ibuprofen on opioid withdrawal in the rat. Anesth Analg. 2000; 91: 417-422.
- Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology. 2011; 114: 1180-1189.
- Bannister K, Sikandar S, Bauer CS, Dolphin AC, Porreca F, Dickenson AH. Pregabalin suppresses spinal neuronal hyperexcitability and visceral hypersensitivity in the absence of peripheral pathophysiology. Anesthesiology. 2011; 115: 144- 152.
- Tumati S, Roeske WR, Largent-Milnes TM, Vanderah TW, Varga EV. Intrathecal PKA-selective siRNA treatment blocks sustained morphine- mediated pain sensitization and antinociceptive tolerance in rats. J Neurosci Methods. 2011; 199: 62-68.
- Célérier E, Simonnet G, Maldonado R. Prevention of fentanyl-induced delayed pronociceptive effects in mice lacking the protein kinase Cgamma gene. Neuropharmacology. 2004; 46: 264-272.
- Esmaeili-Mahani S, Shimokawa N, Javan M, Maghsoudi N, Motamedi F, Koibuchi N, et al. Low-dose morphine induces hyperalgesia through activation of G alphas, protein kinase C, and L-type Ca 2+ channels in rats. J Neurosci Res. 2008; 86: 471-479.
- Sjøgren P, Thunedborg LP, Christrup L, Hansen SH, Franks J. Is development of hyperalgesia, allodynia and myoclonus related to morphine metabolism during long-term administration? Six case histories. Acta Anaesthesiol Scand. 1998; 42: 1070-1075.
- Juni A, Klein G, Kest B. Morphine hyperalgesia in mice is unrelated to opioid activity, analgesia, or tolerance: Evidence for multiple diverse hyperalgesic systems. Brain research. 2006; 1070: 35-37.
- Grastilleur S, Mouledous L, Bedel J, Etcheverry J, Bader M, Girolami JP, et al. Role of kinin B2 receptors in opioid-induced hyperalgesia in in?ammatory pain in mice. Biol Chem. 2013; 394: 361-368.
- Mao J, Chen LL. Gabapentin in pain management. Anesth Analg. 2000; 91: 680-687.
- Van Elstraete AC, Sitbon P, Mazoit JX, Benhamou D. Gabapentin prevents delayed and long-lasting hyperalgesia induced by fentanyl in rats. Anesthesiology. 2008; 108: 484-494.
- Wei X, Wei W. Role of gabapentin in preventing fentanyl- and morphine- withdrawal-induced hyperalgesia in rats. J Anesth. 2012; 26: 236-241.
- Hoffmann O, Plesan A, Wiesenfeld-Hallin Z. Genetic differences in morphine sensitivity, tolerance and withdrawal in rats. Brain Res. 1998; 806: 232-237.
- Liang DY, Liao G, Lighthall GK, Peltz G, Clark DJ. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenet Genomics. 2006; 16: 825-835.
- Liang DY, Liao G, Wang J, Usuka J, Guo Y, Peltz G, et al. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology. 2006; 104: 1054-1062.
- Holtman JR Jr, Wala EP. Characterization of morphine-induced hyperalgesia in male and female rats. Pain. 2005; 114: 62-70.
- Juni A, Klein G, Kowalczyk B, Ragnauth A, Kest B. Sex differences in hyperalgesia during morphine infusion: effect of gonadectomy and estrogen treatment. Neuropharmacology. 2008; 54: 1264-1270.
- Liang DY, Li X, Clark JD. Epigenetic regulation of opioid-induced hyperalgesia, dependence, and tolerance in mice. J Pain. 2013; 14: 36-47.
- Ho A, Dole VP. Pain perception in drug-free and in methadone-maintained human ex-addicts. Proc Soc Exp Biol Med. 1979; 162: 392-395.
- Doverty M, White JM, Somogyi AA, Bochner F, Ali R, Ling W. Hyperalgesic responses in methadone maintenance patients. Pain. 2001; 90: 91-96.
- Schall U, Katta T, Pries E, Klöppel A, Gastpar M. Pain perception of intravenous heroin users on maintenance therapy with levomethadone. Pharmacopsychiatry. 1996; 29: 176-179.
- Petersen KL, Jones B, Segredo V, Dahl JB, Rowbotham MC. Effect of remifentanil on pain and secondary hyperalgesia associated with the heat--capsaicin sensitization model in healthy volunteers. Anesthesiology. 2001; 94: 15-20.
- Luginbühl M, Gerber A, Schnider TW, Petersen-Felix S, Arendt-Nielsen L, Curatolo M. Modulation of remifentanil-induced analgesia, hyperalgesia, and tolerance by small-dose ketamine in humans. Anesth Analg. 2003; 96: 726- 732, table of contents.
- Koppert W, Dern SK, Sittl R, Albrecht S, Schüttler J, Schmelz M. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001; 95: 395-402.v
- Hood DD, Curry R, Eisenach JC. Intravenous remifentanil produces withdrawal hyperalgesia in volunteers with capsaicin-induced hyperalgesia. Anesth Analg. 2003; 97: 810-815.
- Koppert W, Sittl R, Scheuber K, Alsheimer M, Schmelz M, Schüttler J. Differential modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by S-ketamine and clonidine in humans. Anesthesiology. 2003; 99: 152-159.
- Angst MS, Koppert W, Pahl I, Clark DJ, Schmelz M. Short-term infusion of the mu-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain. 2003; 106: 49-57.
- Tröster A, Sittl R, Singler B, Schmelz M, Schüttler J, Koppert W. Modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by parecoxib in humans. Anesthesiology. 2006; 105: 1016-1023.
- Koppert W, Angst M, Alsheimer M, Sittl R, Albrecht S, Schüttler J, et al. Naloxone provokes similar pain facilitation as observed after short-term infusion of remifentanil in humans. Pain. 2003; 106: 91-99.
- Singler B, Tröster A, Manering N, Schüttler J, Koppert W. Modulation of remifentanil-induced postinfusion hyperalgesia by propofol. Anesth Analg. 2007; 104: 1397-1403, table of contents.
- Lenz H, Raeder J, Draegni T, Heyerdahl F, Schmelz M, Stubhaug A. Effects of COX inhibition on experimental pain and hyperalgesia during and after remifentanil infusion in humans. Pain. 2011; 152: 1289-1297.
- Chu LF, Cun T, Ngai LK, Kim JE, Zamora AK, Young CA, et al. Modulation of remifentanil-induced postinfusion hyperalgesia by the β-blocker propranolol in humans. Pain. 2012; 153: 974-981.
- Guignard B, Bossard AE, Coste C, Sessler DI, Lebrault C, Alfonsi P, et al. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology. 2000; 93: 409-417.
- Hansen EG, Duedahl TH, Rømsing J, Hilsted KL, Dahl JB. Intra-operative remifentanil might in?uence pain levels in the immediate post-operative period after major abdominal surgery. Acta anaesthesiologica Scandinavica. 2005; 49: 1464-1470.
- Lee C, Kim YD, Kim JN. Antihyperalgesic effects of dexmedetomidine on high-dose remifentanil-induced hyperalgesia. Korean J Anesthesiol. 2013; 64: 301-307.
- Compton MA. Cold-pressor pain tolerance in opiate and cocaine abusers: correlates of drug type and use status. J Pain Symptom Manage. 1994; 9: 462-473.
- Compton P, Charuvastra VC, Kintaudi K, Ling W. Pain responses in methadone-maintained opioid abusers. J Pain Symptom Manage. 2000; 20: 237-245.
- Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depend. 2001; 63: 139-146.
- Compton P, Canamar CP, Hillhouse M, Ling W. Hyperalgesia in heroin dependent patients and the effects of opioid substitution therapy. J Pain. 2012; 13: 401-409.
- Krishnan S, Salter A, Sullivan T, Gentgall M, White J, Rolan P. Comparison of pain models to detect opioid-induced hyperalgesia. J Pain Res. 2012; 5: 99-106.
- Treister R, Eisenberg E, Lawental E, Pud D. Is opioid-induced hyperalgesia reversible? A study on active and former opioid addicts and drug naïve controls. J Opioid Manag. 2012; 8: 343-349.
- Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain perception: New evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend. 2006; 82: 218-223.
- Doverty M, Somogyi AA, White JM, Bochner F, Beare CH, Menelaou A, et al. Methadone maintenance patients are cross-tolerant to the antinociceptive effects of morphine. Pain. 2001; 93: 155-163.
- Chu LF, D'Arcy N, Brady C, Zamora AK, Young CA, Kim JE, et al. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: a double- blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain. 2012; 153: 1583- 1592.
- Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: a preliminary prospective study. J Pain. 2006; 7: 43-48.
- Compton PA, Ling W, Torrington MA. Lack of effect of chronic dextromethorphan on experimental pain tolerance in methadone-maintained patients. Addict Biol. 2008; 13: 393-402.
- Hong BH, Lee WY, Kim YH, Yoon SH, Lee WH. Effects of intraoperative low dose ketamine on remifentanil-induced hyperalgesia in gynecologic surgery with sevo?urane anesthesia. Korean J Anesthesiol. 2011; 61: 238-243.
- Reznikov I, Pud D, Eisenberg E. Oral opioid administration and hyperalgesia in patients with cancer or chronic nonmalignant pain. Br J Clin Pharmacol. 2005; 60: 311-318.
- Compton P, Kehoe P, Sinha K, Torrington MA, Ling W. Gabapentin improves cold-pressor pain responses in methadone-maintained patients. Drug Alcohol Depend. 2010; 109: 213-219.
- Tompkins DA, Campbell CM. Opioid-induced hyperalgesia: clinically relevant or extraneous research phenomenon? Curr Pain Headache Rep. 2011; 15: 129-136.
- Wanigasekera V, Lee MC, Rogers R, Hu P, Tracey I. Neural correlates of an injury-free model of central sensitization induced by opioid withdrawal in humans. J Neurosci. 2011; 31: 2835-2842.
- McDonnell C, Zaarour C, Hull R, Thalayasingam P, Pehora C, Ahier J, et al. Pre-treatment with morphine does not prevent the development of remifentanil-induced hyperalgesia. Can J Anaesth. 2008; 55: 813-818.
- Crawford MW, Hickey C, Zaarour C, Howard A, Naser B. Development of acute opioid tolerance during infusion of remifentanil for pediatric scoliosis surgery. Anesth Analg. 2006; 102: 1662-1667.
- Mercadante S, Ferrera P, Villari P, Arcuri E. Hyperalgesia: an emerging iatrogenic syndrome. J Pain Symptom Manage. 2003; 26: 769-775.
- Silverman SM. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician. 2009; 12: 679-684.
- Fine PG. Opioid insights:opioid-induced hyperalgesia and opioid rotation. J Pain Palliat Care Pharmacother. 2004; 18: 75-79.