
Special Article - Pain Management
Austin J Anesthesia and Analgesia. 2018; 6(1): 1065.
Buprenorphine for Pain Management in Osteoarthritis Patients Scheduled for Arthroplasty - A Randomized, Double-Blind, Placebo-Controlled Trial
Härkänen L1#, Kokki M1#, Huopio J², Sjövall S³, Auriola S4, Lehtonen M4 and Pergolizzi J5, Kokki H1*
1Department of Anesthesia and Operative Services, Kuopio University Hospital, School of Medicine, University of Eastern Finland, Kuopio, Finland
²Department of Orthopaedic Surgery, Kuopio University Hospital, Kuopio, Finland
³Department of Anesthesia, Satakunta Central Hospital, Pori, Finland
4School of Pharmacy, Faculty of Health Sciences, University of Eastern Finland, Kuopio, Finland
5Department of Analgesic Research, NEMA Research Inc., Naples, Florida, USA
#Both authors are equally contributed
*Corresponding author: Kokki H, Anesthesia and Operative Services, Kuopio University Hospital, Finland
Received: December 14, 2017; Accepted: January 02, 2018; Published: January 09, 2018
Abstract
Background: Preoperative pain is a risk factor for persistent postoperative pain. Low-dose buprenorphine is assumed to have an antihyperalgesic efficiency and thus, started preoperatively may decrease postoperative pain.
Methods: This hypothesis was tested in a double-blind, placebo-controlled clinical trial in 117 osteoarthritis (OA) patients having hip (n=52) or knee (n=65) arthroplasty. Subjects in the buprenorphine-group (n=58) received five 7-day transdermal buprenorphine 5mcg/h patches starting 2 weeks prior to surgery, and subjects in the placebo-group (n=59) similar placebo patches. Numerical pain scores 0-10 were recorded at baseline, during the hospital stay, and four weeks after surgery. The use of analgesics, adverse drug effects and treatment satisfaction were recorded.
Results: In the buprenorphine-group the mean pain score decreased on the preoperative morning compared to pain before patch application (mean difference 0.4; 95% CI, 0.0-0.8; p=0.036). At 48 hours after surgery the average pain score was less in the buprenorphine-group than in the placebo-group (0.8; 0.0-1.5; p=0.043). Both active and placebo patches were well tolerated and satisfaction with treatment was similarly high in both groups.
Conclusion: Low-dose transdermal buprenorphine could be feasible for add-on pain management in OA and arthroplasty patients but further studies are needed to establish optimal dosage.
Keywords: Analgesics; Opioid; Buprenorphine; Arthroplasty; Replacement; Knee; Arthroplasty; Replacement; Hip; Osteoarthritis; Pain; Postoperative
Introduction
Hip and knee osteoarthritis (OA) are the most common joint disease and are associated with significant pain and disability [1]. Early-stage OA treatment consists of pain management and physiotherapy, but as the condition progresses joint replacement often becomes necessary [2,3]. Preoperative pain is a risk factor for postoperative pain [4]. In a study by Gerbershagen et al [5] where data on 4000 arthroplasty patients were available, preoperative pain appeared to be a risk for early postoperative pain and for severe postoperative pain, defined as pain ≥7/10 on a numerical rating scale (NRS). After arthroplasty, 7%-23% of hip replacement and 10%- 34% of knee replacement patients have moderate or severe pain that impacts their daily life for several weeks after surgery [6,7].
Buprenorphine is a highly lipophilic thebaine derivative that has affinity for mu-, kappa-, and delta-opioid peptide receptors (MOP, KOP, DOP) and a low affinity for nociceptin receptors (NOP). It acts as an agonist at the MOP and NOP and as an antagonist at the DOP and KOP. Buprenorphine binds and dissociates from the MOP slowly and thus it has up to a two-fold duration of action and is approximately 30-fold more potent when compared to parenteral morphine [8]. Both antihyperalgesic and antinociceptive efficacy have been proposed [8]. Buprenorphine’s antihyperalgesic action has been demonstrated in a human pain model where hyperalgesia was induced by central sensitization [8].
In this prospective, randomized, double-blind, parallel-group and placebo-controlled clinical trial, we investigated the analgesic efficacy of transdermal buprenorphine initiated before elective total hip arthroscopy (THA) or total knee arthroplasty (TKA) and continued postoperatively for a total of five weeks of treatment. Our hypothesis was that add-on treatment with a transdermal buprenorphine 5mcg/h patch would decrease perioperative pain in patients scheduled for elective THA or TKA.
Methods
The study was conducted in Kuopio University Hospital (KUH), Kuopio and Satakunta Central Hospital (SCH), Pori, Finland between June 2013 and May 2014. The study protocol was approved by the Research Ethics Committee of the Hospital District of Northern Savo, Kuopio, Finland (ref. No. 113/2011), Finnish Medicines Agency (ref. 122//2011) was notified, and it was registered in the European Clinical Trials Database (ref. Eudra CT: 2011-000692-14) and in the ClinicalTrials.gov database (ref. NCT02575664). Both registering institutions approved the study. The study was conducted in accordance with the principles presented in the Declaration of Helsinki. Written informed consent was obtained from each subject.
The primary outcome measures for efficacy were the pain intensity in the preoperative morning after two weeks of patch use; consumption of rescue medication (oxycodone) during the first 24 postoperative hours; pain intensity scores; and analgesics used at four weeks after surgery. Secondary measures were postoperative pain intensity during the first two postoperative days and at four weeks after surgery; length of stay (LOS) in the hospital; patient satisfaction with pain treatment; and suspected adverse drug events (ADEs).
Participants
We recruited adult patients aged 18-75 years scheduled for primary, elective, unilateral THA or TKA due to primary OA; surgery was to be performed under spinal anesthesia. We did not enroll patients with a body-mass index (BMI) less than 18 or over 35kg/m2 or patients with ongoing buprenorphine treatment, hypersensitivity to buprenorphine or to the ingredients of the patch, a history of alcohol or narcotics abuse, treatment with MAO-inhibitors within the last four weeks, renal, hepatic or pulmonary impairment, medically treated constipation within the last three months, and those who were pregnant or lactating. A total of 160 patients were asked to participate and 126 agreed.
Patients were randomly assigned to receive either an active 5mcg/h buprenorphine or a similar placebo patch. There were two sets of patches available, one set for THA and another for TKA patients. Both sets of study patches were consecutively numbered to ensure treatment allocation was concealed from the patients, investigators, treating physicians, and data analysts. The success of blinding was tested by asking patients to guess whether they had an active or placebo patches. Concealment was ended after data analysis.
Study interventions
Patients were enrolled at the preoperative visit two weeks prior to scheduled surgery. At the preoperative visit, the first study patch was applied to those who agreed to participate and patients were instructed to change to a new patch on a different appropriate skin site after seven days. Patients were given both oral and written instructions for the change and care of the patches. Patients were instructed to contact the researchers if they had any questions, concerns, or wanted to discontinue the study treatment. Patients were treated for five weeks, starting two weeks before scheduled surgery and ending two to three weeks after hospital discharge. A total of five patches were used for each patient
Anesthesia
Spinal anesthesia was standardized. Lumbar puncture was performed at the L4/5- or L3/4-interspace. Spinal anesthesia was induced with (levo) bupivacaine 10-15 mg with fentanyl 10-20 mcg.
Surgery
A standardized surgical technique was used. The standard posterior approach was made in every THA. After skin and fascia incisions, external muscles were cut and the posterior capsule was opened. After the hip implants were installed, the posterior muscles were re-inserted in their original positions, and fascia and skin incisions were sutured. In TKA, a tourniquet was applied before the operation. Straight midline skin and medial parapatellar fascial incisions were used in approach. The patella was everted and bone cuts were made by an oscillating saw utilizing intramedullar instrumentation in the femur and extramedullar instrumentation in the tibia. Bone cement was used to fixate the components. The patella was not resurfaced. The fascia and subcutaneous tissues were sutured in layers and the skin was closed with staples. After draping the wound, the tourniquet was deflated.
Postoperative pain management
Postoperative pain was treated according to the hospitals’ standardized protocols. Local infiltration analgesia (LIA) was used for most subjects with TKA (48/51) and many with THA (23/66). All patients were administered paracetamol at a dose of 1g three times daily, with the first postoperative dose administered IV, and by mouth thereafter. If not contraindicated, patients were given an NSAID, first doses IV ketoprofen 50-100 mg, followed by meloxicam 7.5mg x 1-2 by mouth. Controlled-release oxycodone-naloxone (5/2.5mg or 10/5mg) tablets were commenced the first evening, and continued every morning and evening during the hospital stay. Rescue medication was provided as appropriate and could be 2-3 mg IV oxycodone, 5-10 mg subcutaneous oxycodone, or 5-10 mg oxycodone by mouth. At discharge, patients were prescribed paracetamol and NSAIDs for pain control, and tramadol or codeine for rescue analgesia.
Pain assessments
Pain was evaluated on an eleven-point NRS where 0=no pain, and 10=most pain. Patients were asked to rate the least, the average and the most pain at rest, during hip/knee flexion/extension, and while walking during the previous 24 hours. Pain scores were recorded at baseline before patch application (two weeks prior to scheduled surgery), on the preoperative morning, during the recovery room stay, at discharge to the surgical ward, on the first and second postoperative afternoons, at discharge from the hospital, and four weeks after surgery. All suspected ADEs were recorded at these same time points. Satisfaction with pain management was evaluated using a 5-point Likert scale where 1=very satisfied, and 5=very unsatisfied. The efficiency of pain management was rated on a scale 0-10 at baseline, on the preoperative morning, at discharge, and four weeks after surgery. In the preoperative visit patients were asked how severe postoperative pain they expected to have after surgery and how severe postoperative pain they were prepared to accept.
Statistics
Sample size calculation was based on data indicating that patients waiting for arthroplasty would have a mean pain score of 53.3 (standard deviation [SD] 22.6) on a scale 0-100 [10]. To achieve a 10-point decrease in the preoperative pain score, 80 patients per group would be required to achieve a statistical significance (p-value ≤ 0.05) at a power of 0.8.
Data were entered and analysed using IBM SPSS Statistics for Window, Version 23.0. (International Business Machines Corp., Armonk, NY, USA). The normality of the data was assessed for skewness and kurtosis. The intention-to-treat population included all patients who were randomized and had at least one valid aftertreatment efficacy measurement. Binomial and categorical data were analysed using the Chi-Square test. Continuous and nominal data were analysed with an independent sample t-test or the Mann Whitney U-test or related samples t-test, as appropriate. Correlations between patients’ characteristics and analgesic efficacy were analysed using the Pearson correlation coefficient and correlations between preoperative pain and postoperative recovery with Spearman’s correlation coefficient. A two-sided p-value of < 0.05 was considered significant.
Results
A total of 160 patients were asked to participate in the study, 126 were enrolled, of whom 9 were excluded, leaving 117 subjects for the intention-to-treat analysis. Because the study patches expired in May 2014, we did not have time to recruit new patients to replace those who declined or were excluded. The reasons for exclusion in the buprenorphine-group were withdrawn consent (n=3, one of which due to a family emergency) and cancelled surgery (n=1). In the placebo-group the reasons for exclusion were withdrawn consent (n=4, one of which was due to difficulties using transdermal system) and an acute medical condition (n=1). A flow chart is presented in Figure 1.
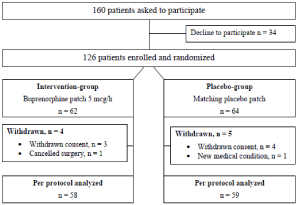
Figure 1: Flow diagram.
The study groups were similar with regard to baseline characteristics; 80 patients were treated at KUH and 37 at SCH (Table 1).
![]()
Buprenorphine
Placebo
N = 58
N = 59
Knee (n=51)
n = 23
n = 28
Female/male
13/10
17/11
Age, yr
65 (6)
63 (6)
Weight, kg
82 (11)
82 (14)
Height, m
169 (7)
171 (10)
BMI, kg/m2
28.7 (3.4)
28.0 (4.4)
ASA, I/II/III
2/11/10
6/8/2014
Preoperative patch wear, d
13.2 (2.3)
12.7 (3.1)
Duration of surgery, min
67 (23)
63 (25)
LIA, yes/no
23/-
25/3
Study hospital, KUH/SCH
16/7
22/6
Hip (n=66)
n = 35
n = 31
Female/male
21/14
23/8
Age, yr
63 (8)
63 (9)
Weight, kg
80 (15)
83 (14)
Height, m
171 (10)
174 (10)
BMI, kg/m2
27.0 (3.4)
27.2 (2.9)
ASA, I/II/III
7/19/9
7/176/7
Preoperative patch wear, d
14.0 (3.7)
13.4 (2.1)
Duration of surgery, min
58 (23)
66 (27)
LIA, yes/no
25-Oct
13/18
Study hospital, KUH/SCH
25/10
18/13
BMI: Body Mass Index; ASA: American Society of Anesthesiologists physical status; LIA: Local Infiltration Analgesia; KUH: Kuopio University Hospital; SCH: Satakunta Central Hospital
Table 1: Baseline characteristics and surgical data. Data are mean (standard deviation) or number of cases.
Preoperative visit
At baseline, there were no significant differences between the groups with regard to pain scores, satisfaction or ADEs. Hip OA subjects reported more severe pain than patients with knee OA (6.7 (1.8) vs. 5.6 (2.4), respectively, p=0.014) (Figure 2). Nine subjects reported 11 ADEs, the most commonly reported of which were somnolence (n=3), constipation (n=2) and increased sweating (n=2). Most suspected ADEs were associated with the opioid analgesics.
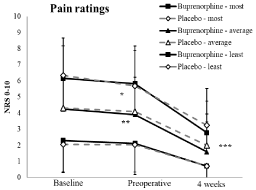
Figure 2: Pain scores in the two study groups at baseline before study patch
application, in preoperative morning after two weeks of wearing the patch and
at 4 weeks after surgery as its average, least and most during the previous
24 hours. Data are mean and standard deviation. NRS = numerical rating
scale. *p = 0.013 in the placebo-group compared to baseline, **p = 0.032 in
the buprenorphine-group compared to baseline, ***p < 0.001 compared to
baseline and preoperative in both groups.
Patients in both groups said they expected early postoperative pain of 7.8 (1.6) vs. 7.7 (1.7) in the buprenorphine- and placebogroup, respectively. However, patients in both groups were prepared to accept significantly lower pain levels of 5.0 (1.6) and 5.2 (1.8), respectively (p<0.001 compared to the “expected pain”).
Preoperative morning
In the preoperative morning, there were no significant differences between the groups with regards to pain scores, pain right now was 2.9 (1.8) vs. 3.3 (2.4) in the buprenorphine- and placebo-groups, respectively (mean difference, 0.5; 95% confidence interval [CI], -0.3- 1.3; p=0.26).Buprenorphine patients reported their average pain score during the previous 24 hours as significantly lower than their pain scores at baseline (mean difference, 0.4; 95% CI, 0.0-0.8; p=0.032). In the placebo-group the most severe pain decreased in the preoperative morning compared to that before patch application (mean difference, 0.6; 95% CI, 0.1-1.2; p=0.013), see Figure 2. Significantly fewer buprenorphine patients had taken analgesics during the last 24h on the preoperative morning than before patch application (p=0.001), which suggests that the active patches conferred either an analgesic or antihyperalgesic effect. No such difference was observed in placebo patients (Table 2).
![]()
Any
Paracetamol
NSAIDs
Opioids
Had in use before surgery
Buprenorphine, n=58
51 (88%)
35 (60%)
37 (64%)
11 (19%)
Placebo-group, n=59
51 (86%)
31 (53%)
36 (61%)
11 (24%)
Had used during previous 24h
At baseline before study patch application
Buprenorphine, n=58
37 (64%)
21 (36%)
23 (40%)
8 (14%)
Placebo-group, n=59
39 (66%)
21 (36%)
26 (44%)
6 (10%)
Pre-operative morning
Buprenorphine, n=58
27 (47%)
14 (24%)*
15 (26%)**
6 (10%)**
Placebo-group, n=59
34 (58%)
25 (42%)**
14 (24%)
7 (12%)**
Post-operatively at one month
Buprenorphine, n=58
47 (78%)
39 (67%)
24 (41%)***
12 (21%)
Placebo-group, n=59
50 (85%)
37 (63%)
39 (66%)
20 (34%)
* p = 0.002 vs. baseline; ** p < 0.001 vs. baseline; *** p = 0.007 between the two groups.
Table 2: The use of analgesics during the previous 24 h. Data are number of cases (percentage).
Blinding performed sufficiently; two-thirds in both groups could correctly guess their group assignment (p=0.074).
After two weeks of the add-on treatment, there was no significant difference between the groups with regards to satisfaction; 4 buprenorphine and 7 placebo patients reported being dissatisfied with their pain management. In the buprenorphine-group 28 patients had a total of 58 ADEs and in the placebo-group 23 patients reported a total of 47 ADEs (Table 3).
![]()
Buprenorphine
n=58
Placebo
n=59
Total number of subjects with ADE
28
23
Total number of ADE
58
s47
Pruritus
12
5
Fatigue
8
2
Tiredness
2
2
Dizziness
2
2
Confusion
-
4
Sleepiness
-
1
Anorexia
1
-
Depression
-
1
Nausea
8
4
Constipation
7
7
Diarrhoea
2
1
Abdominal pain
1
1
Flatulence
-
2
Muscle pain
1
-
Tremor/perioral tremor
1-Jan
-
Headache
3
1
Mouth dry
1
4
Sweating increased
2
3
Pollakiuria
-
1
Influenza-like symptoms
1
-
Therapeutic response decreased
-
4
Application site pain/rash/redness/vesicle
1/1/2/1
-/1/1/-
Table 3: Suspected adverse drug effects (ADE) in the preoperative morning after two weeks use of study patches. Data are number of cases.
In the hospital
In the first postoperative morning, pain scores were similar in the two groups. Patients had realistic expectations of the outcome, both the most pain at rest, 7.4 (2.2) and the most dynamic pain 7.8 (2.0), had a positive correlation with the preoperative assumption, r=0.32; p=0.001 and r=0.33; p=0.001, respectively, see Figure 3.
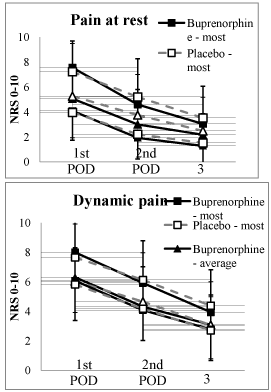
Figure 3: Pain scores in the two study groups in hospital as its average
and most during the previous 24 hours and right now. Data are mean and
standard deviation. NRS = numerical rating scale. POD = postoperative
morning. *p = 0.043 the average pain during the last 24 hours buprenorphinegroup
vs. placebo-group.
At 48 hours after surgery buprenorphine patients reported less pain than placebo patients; the difference was significant in the average pain score during the previous 24 hours at rest, 3.0 (1.8) vs. 3.9 (2.1) (mean difference, 0.9; 95% CI, 0.2-1.6; Cohen’s d 0.45; p=0.014) (Figure 3).
The cumulative oxycodone consumption for rescue analgesia was similar in the two groups, 61 (22) mg and 103 (33) mg for 24 and 48 hours in the buprenorphine-group compared to 65 (29) and 114 (46) mg for the placebo-group.
In THA patients those in the buprenorphine-group used an average 21mg less rescue oxycodone, 98 (34) mg for buprenorphine patients versus 119 (54) for placebo patients (95% CI -2-44 mg; p=0.074). In a post-hoc analysis it was revealed that among hip OA patients those 23 who had LIA reported less pain at rest during the first hours after surgery, 1.4 (1.2), than those 43 without LIA, 3.1 (2.1) (p=0.001) but patients with LIA needed more oxycodone for rescue analgesia during the 25-48 postoperative hours than those without LIA, 47 (20) mg vs. 34 (25) mg, respectively.
In the buprenorphine-group 24/57 patients had 37 adverse effects and in the placebo-group 33/58 subjects had 49 adverse effects. Postoperative nausea and/or vomiting and constipation were most common, but no serious or unexpected ADEs were reported.
At discharge, the pain scores were similar in the two study groups, and 9/51 buprenorphine patients and 15/55 placebo patients reported moderate to severe pain at rest, and four and eight subjects had severe pain on movement; (Figure 3). The length of stay was similar in the two groups. However, in the THA subjects with buprenorphine the mean LOS was 3.2 (0.8) days compared to 3.6 (0.9) days in subjects with placebo patches (p=0.087).
Most subjects were satisfied or very satisfied with the pain management in the hospital; one THA patients in the placebo-group was dissatisfied.
Four weeks after surgery
At four weeks after surgery there were no significant differences between the two groups in pain ratings. The numeric values for average pain were 1.6 (1.8) vs. 2.1 (2.2) (mean difference 0.6; 95% CI -0.2-1.3; p=0.14) and highest pain scores 2.7 (1.9) vs. 3.4 (2.4) (mean difference 0.6; 95% CI -0.2-1.4; p=0.12) for the buprenorphine- and placebo-group, respectively. The pain scores were significantly lower in both groups compared to baseline and the preoperative morning (p<0.001). However, in the buprenorphine-group 15 subjects and in the placebo-group 21 subjects (p=0.28) reported high pain scores at rest (>3) and six subjects in the buprenorphine-group and 13 subjects in the placebo-group (p=0.087), respectively, reported significant pain upon walking. Dynamic pain that was greater than that experienced at baseline was reported by 4/56 and 8/54 patients in the buprenorphine- and placebo-groups, respectively (p=0.20). Most patients reporting significant pain at rest (26/36) or while walking (14/19) had undergone TKA (Figure 2).
At four weeks, all TKA patients except one were still taking analgesics, but among THA subjects, 13/35 and 7/30 in the buprenorphine- and placebo-groups, respectively, had discontinued analgesics (p=0.23). The buprenorphine patients used significantly less NSAIDs (n=22) than placebo patients (n=38, p=0.004) (Table 2).
At four weeks after surgery, there were no significant difference between the groups with regard to satisfaction with their pain management and ADEs. Most buprenorphine (50/58) and placebo (49/59) patients reported being satisfied with their pain control.
Correlation of preoperative pain with postoperative recovery
In the placebo-group there were some moderate positive correlations between patients’ preoperative pain and postoperative recovery and the use of rescue analgesics but no such correlations were found in the buprenorphine-group (Table 4).
![]()
Pain before first patch application
Postoperative recovery
Pain at 4 h
Pain at 24 h
Pain at 48 h
Pain at 4 wk
Oxycodone consumption first 48 h
Placebo
n = 59
Pain at rest
r = 0.13
p = 0.34
r = 0.11
p = 0.42
r = 0.4
p = 0.004
r = 0.28
p = 1.0
r = 0.2
p = 0.16
Dynamic pain
r = 0.23
p = 0.1
r = 0.18
p = 0.2
r = 0.25
p = 0.075
r = -0.07
p = 0.64
r = 0.32
p = 0.019
Buprenorphine
n = 58
Pain at rest
r = 0.25
p = 0.07
r = 0.26
p = 0.06
r = 0.17
p = 0.24
r = 0.24
p = 0.07
r = -0.01
p = 0.94
Dynamic pain
r = 0.29
p = 0.035
r = 0.2
p = 0.14
r = 0.07
p = 0.61
r < -0.08
p = 0.56
r = 0.04
p = 0.8
Table 4: Correlations of preoperative pain with postoperative recovery. Data are Spearman’s r and 2-sided p-value.
Complaints associated with patch use
Adherence to study patch use was high, a total of 110 subjects used all five study patches. Four buprenorphine and three placebo patients wanted to stop after two patches, three due to ADEs (two buprenorphine patients and one placebo patient), and one buprenorphine patient due to lack of efficacy. Three patients did not give any specific reason for early termination.
Eight buprenorphine and 13 placebo patients reported application-site ADEs. The most commonly reported patch-related symptoms were application site pruritus (n=7), rash (n=5), pain (n=2), and redness (n=2). Two patients developed skin ulcers and one reported local oedema. In one patient, one of the patches detached. One hypersensitivity reaction occurred in a placebo patient; she was the only patient who consulted a physician about a suspected ADE.
Discussion
Previously, buprenorphine had demonstrated a significant and long-lasting antihyperalgesic action in a human pain model where hyperalgesia was induced by transcutaneous stimulation [9]. Our double-blind, placebo-controlled study suggests this could also be the case for both OA and postoperative pain. Compared to placebo patients, buprenorphine patients had lower pain scored on the second postoperative morning and consumed less analgesics both at two weeks wearing the patches, and at four weeks after surgery. This supports the hypothesis that buprenorphine has antihyperalgesic action. Moreover, fewer patients, four in the buprenorphine-group compared to eight patients in the placebo-group, reported dynamic pain more severe than at baseline two weeks after the last patch was removed.
Our study is novel in that we demonstrated for the first time these effects in OA patients following orthopaedic surgery. OA pain is characterised by peripheral and central sensitization [6]. Recent studies have shown that buprenorphine may intensify the effect of the inhibitory descending pain control system [9,11,12]. Our results, however, support earlier work suggesting that, as a single compound, buprenorphine’s analgesic efficacy at low transdermal doses in OA pain is relatively small at best. In these earlier positive studies transdermal buprenorphine had been titrated to effect and used at doses up to 20mcg/h [8]. In the present study, a standardized strength of 5mcg/h buprenorphine was used.
Patches were well tolerated and no serious or unexpected ADEs were reported. The ADE incidence as well as proportion of subjects with suspected ADEs is known to be similar in both active and placebo groups [13]. The rate of withdrawals due to suspected ADEs was low. A total of nine patients, five with active and four with placebo patches discontinued the treatment due to AEs that were suspected to be related to the treatment. These results are similar or less than those reported earlier indicating patients’ high adherence in the present study [14]. Most patients were taking concomitant medication and had underlying diseases, thus, some ADEs may not have been related to the study patch or the active ingredient. Nine of those 110 subjects who completed the study reported application site reactions, but they did not need to stop the treatment as their symptoms were mild and transient. Subsequent patches placed at new application sites were well tolerated.
Our study has several strengths. First, the blinding performed relatively well. Secondly, the Hawthorne effect is unlikely as the patients in the present study reported similar or higher pain scores than those in a previous study on THA patients (Sep/2012- Apr/2013) [15] and a subsequent study TKA patients (May/2014- September/2014 data on file) from a same centre.
One of the limitations of the present study was a relatively small sample size. Our aim was to enroll 160 subjects, but some declined to participate and eight subjects were excluded from both groups, leaving 117 subjects for the intention-to-treat analysis. Unfortunately, the patches used for our study expired in May 2014 and thus, we did not have enough patches for new subjects to replace those who declined or withdrew. However, it was considered a clinically meaningful difference that four in the buprenorphine-group compared to eight in the placebo-group had more dynamic pain at four weeks after surgery than they had had before surgery. Moreover, the increase in pain severity was less in the buprenorphine-group, three patients reported an increase of pain score of one, and one patient reporting going from 0/10 to 5/10 compared to the placebo-group, where two patients had an increase of one point, two with two points, three with three points and one patient went from 2/10 to 8/10. The dose of buprenorphine may also have played a role in our findings. Initially, we planned to use 10mcg/h patch based on the published data about transdermal buprenorphine [16]. This plan changed based on the summary of prescribing information, which stated opioid-naïve patients should be initiated with a 5mcg/h buprenorphine patch and the fact that matching placebo patches were available only for 5mcg/h active patches. The decrease in pain score in the active-treatment group was less than the assumption used in the sample size calculation and congruent, although lower, in the placebo-group. Unfortunately, patient adherence was suboptimal. Although patients were provided with both oral and written instructions on the use of study patches as an add-on treatment to regular analgesics, only a few patients continued their regular analgesics as before. More buprenorphine than placebo patients discontinued or reduced their use of other analgesics while wearing the study patch. This may have obscured the potentially synergistic analgesic effect of buprenorphine anticipated in concomitant use with nonopioid analgesics.
Conclusion
Our data support the findings from previous studies indicating that low-dose transdermal buprenorphine could be feasible for addon pain management in OA and arthroplasty patients. However, in most patients a perioperative 5mcg/h patch offers, at best, relatively modest analgesic benefit, and thus further studies are needed to establish optimal dosage.
Compliance with Ethical Standards
Funding: This study was funded by a governmental research grant number 5070244 from the Hospital District of Northern Savo, Kuopio, Finland. Mundipharma Oy (Vantaa, Finland) provided consecutively numbered active and placebo patches, but the company was not otherwise involved in the study.
Conflicts of Interest
Lasse Härkänen, Merja Kokki, Jukka Huopio, Sari Sjövall, Seppo Auriola, Marko Lehtonen, Joseph Pergolizzi and Hannu Kokki have no conflicts of interest that are relevant to this work.
Ethical approval: All procedures were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments.
Informed consent: Informed consent was obtained from all individual participants included in the study.
Contributions
L.H: study design, data analysis, writing of the first draft of the manuscript, critical evaluation of the intellectual content of the final version; M.K: study design, patient recruitment, data collection, data analysis and interpretation of data, critical evaluation of the intellectual content of the final version; J.H: patient recruitment, data collection, analysis and interpretation of data, critical evaluation of the intellectual content of the manuscript; S.S: study design, patient recruitment, data collection, critical evaluation of the intellectual content of the manuscript; S.A: study design, critical evaluation of the intellectual content of the manuscript; M.L: study design, critical evaluation of the intellectual content of the manuscript; J.P: interpretation of data, critical evaluation of the intellectual content of the manuscript; H.K: principal investigator, study design, patient recruitment, data collection, data analysis, analysis and interpretation of data, writing the manuscript, final approval of the version to be published.
References
- Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, Carr AJ. Osteoarthritis. Lancet. 2015; 386: 376-387.
- Carr AJ, Robertsson O, Graves S, Price AJ, Arden NK, Judge A, Beard DJ. Knee replacement. Lancet. 2012; 379: 1331-1340.
- Pivec R, Johnson AJ, Mears SC, Mont MA. Hip arthroplasty. Lancet. 2012; 380: 1768-1777.
- Kehlet H, Jensen TS, Woolf CL. Persistent postsurgical pain: Risk factors and prevention. Lancet. 2006; 367:1618-1625.
- Gerbershagen HJ, Pogatzki-Zahn E, Aduckathil S, Peelen LM, Kappen TH, van Wijck AJ, et al. Procedure-specific risk factor analysis for the development of severe postoperative pain. Anesthesiology. 2014; 120: 1237-1245.
- Beswick AD, Wylde V, Gooberman-Hill R, Blom A, Dieppe P. What proportion of patients report long-term pain after total hip or knee replacement for osteoarthritis? A systematic review of prospective studies in unselected patients. BMJ Open. 2012; 2: e000435.
- Ip HY, Abrishami A, Peng PW, Wong J, Chung F. Predictors of postoperative pain and analgesic consumption: A qualitative systematic review. Anesthesiology. 2009; 111: 657-677.
- Plosker GL. Buprenorphine 5, 10 and 20 μg/h transdermal patch: A review of its use in the management of chronic non-malignant pain. Drugs. 2011; 71: 2491-2509.
- Koppert W, Ihmsen H, Körber N, Wehrfritz A, Sittl R, Schmelz M, Schüttler J. Different profiles of buprenorphine-induced analgesia and antihyperalgesia in a human pain model. Pain. 2005; 118: 15-22.
- Gossec L, Paternotte S, Maillefert JF, Combescure C, Conaghan PG, Davis AM, Gunther KP, et al. The role of pain and functional impairment in the decision to recommend total joint replacement in hip and knee osteoarthritis: an international cross-sectional study of 1909 patients. Report of the OARSIOMERACT Task Force on total joint replacement. Osteoarthritis Cartilage. 2011; 19: 147-154.
- Ravn P, Secher EL, Skram U, Therkildsen T, Christrup LL, Werner MU. Morphine- and buprenorphine-induced analgesia and antihyperalgesia in a human inflammatory pain model: A double-blind, randomized, placebocontrolled, five-arm crossover study. J Pain Res. 2013; 6: 23-38.
- Tröster A, Ihmsen H, Singler B, Filitz J, Koppert W. Interaction of fentanyl and buprenorphine in an experimental model of pain and central sensitization in human volunteers. Clin J Pain. 2012; 28: 705-711.
- Sorge J, Sittl R. Transdermal buprenorphine in the treatment of chronic pain: Results of a phase III, multicenter, randomized, double-blind, placebocontrolled study. Clin Ther. 2004; 26: 1808-1820.
- Huang P, Kehner GB, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: Norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther. 2001; 297: 688-695.
- Piirainen A, Kokki M, Hautajärvi H, Lehtonen M, Miettinen H, Pulkki K, et al. The cerebrospinal fluid distribution of postoperatively administred dexketoprofen and etoricoxib and their effect on pain and inflammatory markers in patients undergoing hip arthroplasty. Clin Drug Investig. 2016; 36: 545-555.
- Al-Tawil N, Odar-Cederlöf I, Berggren AC, Johnson HE, Persson J. Pharmacokinetics of transdermal buprenorphine patch in the elderly. Eur J Clin Pharmacol. 2013; 69: 143-149.