
Case Report
Austin Anesthesiol. 2017; 1(1).
Anaesthetic Management of Extramedullary Pheochromocytoma
Kar SK*, Ganguly T, Dasgupta CS and Goswami A
Department of Cardiac Anesthesiology, Institute of Postgraduate Medical Education & Research, India
*Corresponding author: Sandeep Kumar Kar, Department of Cardiac Anaesthesiology Institute of Postgraduate Medical Education & Research, Kolkata, India
Received: March 06, 2017; Accepted: May 18, 2017; Published: May 25, 2017
Abstract
Pheochromocytomas are catecholamine-secreting tumors of chromaffin tissue and are a rare cause of hypertension. Less than 0.1% of the hypertensive population has a pheochromocytoma. The hypertension caused by these tumors is usually curable. Surgery on a patient with an unrecognized pheochromocytoma can be fatal; similarly the administration of β-adrenergic-blocking drugs can have untoward side effects. These tumors can be associated with other potentially fatal but curable diseases.
Introduction
The high incidence of pheochromocytoma in families as a primary disease, in association with multiple endocrine neoplasia or other familial diseases such as Von Hippel Lindau syndrome and neurofibromatosis I, indicates the need for genetic counseling [1-4]. Approximately 16% of pheochromocytomas will be associated with other endocrine disorders, such as multiple endocrine syndromes [2], which is comprised of medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia.
The Case:
- Twelve years old girl.
- H/O repeated attacks of dyspnoea, palpitation, headache, diaphoresis and cold clammy skin.
- On examination her NIBP was 182/126 mm Hg.
- CT of chest revealed large well defined thoracoabdominal SOL in right paravertebral region of size 50 x 64 x 87 mm.
- On CT abdomen the mass in right paravertebral region extended from T8-to T11
- Twenty four hour urinary VMA was 52.57 mg.
- So, it was diagnosed to be EXTRAMEDULLARY PHEOCHROMOCYTOMA
Pathophysiology
The incidence of pheochromocytoma as a benign tumor in one of the adrenal glands is 80%. Twenty percent are extra-adrenal, with half located below the diaphragm in areas such as along the aorta, near the urinary bladder, and in the organ of Zuckerkandl; the other half is located above the diaphragm in areas along the aorta, in the lungs or heart, or in the neck or carotid bodies. Ten percent occur in children. In nonfamilial disease, the classical teaching is that 10% of patients have bilateral adrenal tumors and 10% have multiple extra-adrenal tumors; however, in familial disease, more than 80% are bilateral or in multiple sites. The incidence of a malignant pheochromocytoma is 10%. The occurrence of a pheochromocytoma is evenly distributed between the sexes and can occur at any age, although the peak incidence is between the fourth and sixth decades.
Catecholamine secretion is responsible for the signs and symptoms of a pheochromocytoma [5]. It is unusual that a tumor will grow large enough or be so invasive as to interfere with the function of the surrounding organs. The manifestations of a pheochromocytoma are primarily the result of the excessive secretion of norepinephrine, epinephrine, and dopamine [6]. The most common combination is predominantly norepinehrine and epinephrine.
Some tumors secrete only norepinehrine, but <10% secrete only epinephrine. Dopamine and its metabolite are more likely to be significantly elevated in children with a pheochromocytoma.
The etiology for the increased production and secretion of catecholamines is not clear. It is conceivable that the negative feedback mechanism of norepinephrine on tyrosine hydroxylase is altered so that sensitivity to feedback is decreased, or perhaps metabolism or release is so rapid that feedback does not transpire in the usual manner. Small tumors tend to secrete high levels of circulating catecholamines. With intracellular metabolism being more prevalent in large tumors, high levels of metabolites tend to be released, and free catecholamine secretion is reduced.
Approximately 50% of patients with a pheochromocytoma have sustained hypertension, 45% are normotensive with paroxysms of hypertension, and 5% are normotensive or even hypotensive [4,6]. These differences, in part, relate to the patterns of catecholamine secretion; bursts produce hypertensive episodes. Patients with sustained hypertension and some normotensive patients can have high or normal levels of norepinephrine. How sustained levels of high norepinephrine concentrations result in persistent hypertension is not understood. However, if elevated catecholamine secretion persists, α- and β-receptors may become desensitized, or even down regulated. Homodynamic mechanisms will no longer respond to elevated levels of norepinephrine, and blood pressure will normalize.
Catecholamine levels and blood pressure do not usually correlate well, but a significant change in catecholamine concentration will elicit a blood pressure response. Patients with the rare, exclusively epinephrine producing tumors can present with normotension, or even hypotension, secondary to the vasodilating properties of epinephrine. Orthostatic hypotension is another result of the ganglionic-blocking activity of excessive amounts of catecholamines (Figure 1- 3).
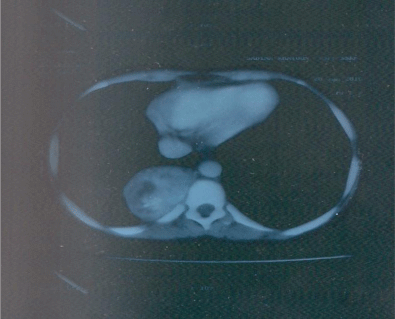
Figure 1: CT of chest revealed large well defined thoracoabdominal SOL
in right Para vertebral region of size 50 x 64 x 87 mm.a vertebral region
extended from T8-to T11.
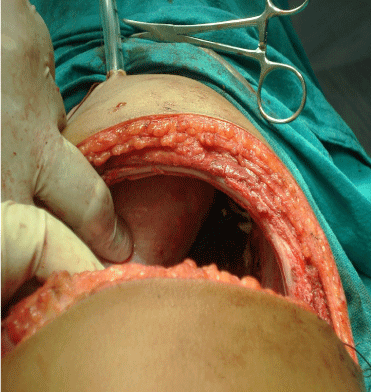
Figure 2: Midline thoracotomy for accesses to the tumor.
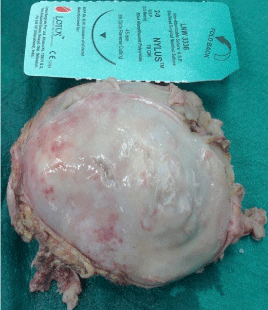
Figure 3: The resected tumour after midline thoracotomy.
Clinical Presentation
The classic triad of symptoms in patients with a pheochromocytoma consists of episodic headache, sweating, and tachycardia, all of which are largely due to the pharmacologic effects of the catecholamines secreted from these tumors [4,7].
Other signs and symptoms include pallor, orthostatic hypotension, visual blurring, papilledema, weight loss, polyuria, polydipsia, increased erythrocyte sedimentation rate, hyperglycemia, psychiatric disorders, and, rarely, secondary erythropoesis consistent with overproduction of erythropoietin. The abnormalities in carbohydrate metabolism are directly related to increases in catecholamine production; these changes resolve after adrenalectomy. There are two rare presentations of a pheochromocytoma: Episodic hypotension in patients with tumors that secrete only epinephrine and rapid cyclic fluctuations of hypertension and hypotension [7-9]. The latter group of patients can be treated by fluid repletion and α-adrenergic antagonists.
These patients can exhibit significant baseline Electrocardiographic (ECG) changes due to the toxic effects exerted on the myocardium by the excessively high levels of catecholamines. Patients may also present with chest pain and ECG changes suspicious for ischemia.
Despite striking repolarization changes, many patients who proceed to coronary angiography preoperatively are found to have no obstruction of their coronary arteries. Anecdotal reports suggest ECG changes to be a manifestation of toxic myocarditis. In addition to the ECG changes mentioned, many patients with a pheochromocytoma are noted to have a long QTc interval which may predispose to ventricular arrhythmias [10,11]. After removal of the pheochromocytoma, the QTc intervals tend to normalize.
An elevated temperature more commonly reflects a catecholamine mediated increase in the metabolic rate and diminished heat dissipation secondary to vasoconstriction. Polyuria is an occasional finding, and rhabdomyolysis with resultant myoglobinuric renal failure may result from extreme vasoconstriction and ensuing muscle ischemia.
Diagnosis
The diagnosis of pheochromocytoma is usually suggested by the history in a symptomatic patient or the family history in a patient with familial disease, and can usually be confirmed by measurements of urinary and plasma catecholamines and metabolites and radiological tests.
Specific Tests for Diagnosing Pheochromocytoma
The measurement of urinary catecholamines and metabolites (preferably metanephrines rather than vinyl mandelic acid) is a useful screening test. Urinary metanephrine excretion above 1.2 mg/d is highly suggestive of a pheochromocytoma, and normal levels on three 24-h collections of metanephrines virtually exclude the diagnosis [12,13]. Many drugs (such as labetolol and buspirone) and dietary vanilla can interfere with these tests. Urinary metanephrine and catecholamine secretion does not vary as a function of age or sex in normal subjects.
Resting plasma catecholamines are elevated.
Plasma chromogranin A (a storage vesicle protein that is co-stored and co-secreted with catecholamines) is elevated.
Clonidine suppression and glucagon stimulation tests may be appropriate, but should be performed in specialized centers.
Computer tomography scans, initially of the abdomen, are helpful to localize the tumors which are often large [14].
Magnetic resonance imaging usually shows the lesion clearly [15].
Scanning with [13,11] metaiodobenzylguanidine produces specific uptake in sites of sympathetic activity, with approximately 90% success; it is especially useful with extra-adrenal tumors [16].
Preparing the patient for surgical removal
pheochromocytoma resection is widely considered to be among the most challenging in anesthetic practice, with the primary goal being the delivery of an anesthetic which provides stable hemodynamic in the face of catecholamine surges (especially at laryngoscopy, peritoneal insufflation, surgical stimulation, and tumor handling) followed by the opposite scenario following tumor ligation. Careful planning and meticulous technique are essential, in addition to close communication with the surgical team
Preparing the patient for surgical removal of a pheochromocytoma entails the institution of α- and β-adrenergic blockade. The reduction in perioperative mortality rates, from as high as 45% to as low as 0% to 3% after the pheochromocytoma is excised, has encouraged the administration of α-antagonists for preoperative therapy which is usually initiated once the diagnosis is established.
To date, no one has investigated the optimal duration of preoperative phenoxybenzamine therapy. Most patients require treatment for 10 to 14 days, as determined by the time needed for stabilization of blood pressure and amelioration of symptoms. If the patient does not complain of nasal stuffiness, he or she is not ready for surgery. Because pheochromocytomas spread slowly, little is lost by waiting until the patient's preoperative condition has been optimized by means of medical therapy. The following criteria for an optimal preoperative condition are recommended:
- No “in-hospital” blood pressure reading higher than 160/90 mm Hg should be evident for 24 hours before surgery. I normally measure the blood pressure in each patient (as an outpatient) every minute for an hour in a recovery room setting during preoperative visits. This setting is most stressful to the medically naive and thus a good test of inhibition of responses to sympathetic stimulation. If no blood pressure reading higher than 160/90 mm Hg is recorded, the patient is scheduled for surgery, assuming the following three criteria are also met.
- Orthostatic hypotension, with readings above 80/45 mm Hg, should be present.
- The electrocardiogram should be free of ST-T abnormalities for at least a week; if abnormalities are persistent, two-dimensional echocardiography should reveal no evidence of global or regional dysfunction that cannot be attributed to a permanent deficit.
- The patient should have no more than one premature ventricular contraction every 5 minutes.
Phenoxybenzamine, a long-acting (24 to 48 hours), noncompetitive, presynaptic, α-adrenergic antagonist, is typically initiated at least 7 days (usually for 2 to 4 weeks) before surgery and added incrementally until the blood pressure is controlled and paroxysms disappear. The initial dose is usually 10 mg every 12 hours. Most patients require between 80 and 200 mg per day. The combination of α-adrenergic receptor blockade and a liberal salt intake will restore the contracted plasma volume towards normal [17]. Selective α-blockers, such as doxazosin, prazosin and terazosin, have also been used effectively, but their role in preoperative management may be limited to the treatment of individual paroxysms.
Nitroprusside, calcium channel blockers (where it was found that treatment extended postoperatively with calcium channel blockers is also effective in treating the clinical manifestations of catecholamine-induced myocarditis and coronary vasospam), [18] and angiotensin-converting enzyme inhibitors all can be used to reduce blood pressure in these patients. Intraoperatively, nitroprusside is beneficial in the treatment of hypertensive crises.
β-adrenergic blockade is initiated more than 3 days before surgery in patients with persistent tachycardia or reflex tachycardia related to the initiation of α-blockade, or in those having arrhythmias. It is important to initiate α-blockade before β-blockade to avoid the situation of unopposed α-agonism whereby the patient suffers from intense vasoconstriction from the α-adrenergic excess and is at risk for extreme hypertension as well as a dramatic increase in myocardial workload. Low doses often suffice; a reasonable starting dose is 10 mg propranolol, three to four times per day, and is increased as needed to control the pulse rate. Labetolol, a β-adrenergic antagonist with some α-blocking activity, is effective as a second-line medication, but can increase the blood pressure if used alone, presumably because of its β-blocking effect being much more pronounced than its α-blockade. The pharmacologic adrenergic blockade helps to blunt the intense surges in blood pressure that occur during surgery and tumor manipulation.
Intravascular volume is also contracted in patients with a pheochromocytoma. This is manifested by hemo concentration and orthostatic changes in blood pressure. α-adrenergic-mediated vasoconstriction, and possibly altered capillary permeability, is thought to be responsible for these findings. Preoperatively, α-adrenergic blockade enables the patient to replete intravascular volume. The presence or absence of orthostasis and changes in hematocrit should be assessed at the time of the preoperative visit. Despite the fact that hypotension commonly occurs after vascular isolation of the tumor, most clinicians continue α-blockade up until the morning of the surgery.
Preoperative treatment with α-methyl-para-tyrosine results in depletion of tumor catecholamine stores caused by the competitive inhibition of tyrosine hydroxylase, and decreases both blood pressure lability and intraoperative blood loss [3]. This medication is currently reserved for patients with metastatic disease or for situations in which surgery is contraindicated and long-term medical therapy is needed.
Cardiovascular Monitoring and Management
Potentially life-threatening fluctuations in blood pressure indicate the need for direct arterial pressure monitoring, and large intraoperative fluid shifts underscore the importance of excellent intravenous access and urinary output monitoring.
Young patients with healthy hearts may need only central venous pressure monitoring, whereas those with a history of cardiac disease, including catecholamine-induced cardiomyopathy, may benefit from transesophageal echocardiography or a pulmonary artery catheter.
Endotracheal intubation should be attempted only after a deep level of anesthesia is attained. Intraoperative hypertension can be effectively treated with phentolamine (a short-acting α-adrenergic antagonist that may be given as an intravenous bolus of 2 to 5 mg or by continuous infusion), nitroprusside, or nicardipine titrated to effect. Other agents that can be used include nitroglycerin, fenoldopam, [19] diltiazem, prostaglandin E1, and magnesium sulfate (as an infusion) [20].
Drugs to be avoided and problems in laparoscopic removal
Anesthetic drugs and techniques that stimulate the sympathetic nervous system, such as ephedrine, ketamine, and hypercarbia; potentiate the arrhythmogenic effects of catecholamines (e.g., halothane); inhibit the parasympathetic nervous system (e.g., pancuronium); or release histamine (e.g., morphine sulfate, atracurium) may precipitate hypertension and are best avoided.
During laparoscopic surgery, creation of the pneumoperitoneum may trigger the release of catecholamines and huge fluctuation in hemodynamics that can be controlled with a vasodilator. Interestingly, the hemodynamic changes are typically very similar when pheochromocytomas are resected laparoscopically compared to when they are removed through laparotomy. [21,22] Blood loss and length of stay are less with the laparoscopic than the open procedure [21]. Anesthesia is often maintained with an opioid analgesic and a potent volatile agent. If a potent volatile anesthetic is used solely to maintain anesthesia, the drop in the blood pressure can be dangerous after tumor resection.
After tumor ligation and resection, the drop in blood pressure can be dangerously abrupt; however, this can be anticipated through close communication with the surgical team. Many patients require a vasoconstrictor (e.g., phenylephrine infusion) to support the blood pressure for a period of hours after the tumor is removed. If postoperative hypertension ensues, it may indicate the presence of occult tumor or volume overload. After successful surgery, catecholamine excretion returns to normal in about two weeks.
In this case the following process of induction and maintenance of anesthesia was done
- Premedicated with tab nitrazepam 5mg on H.S & M.O.S
- Preinduction IVI 500 ml R.L.
- Induction:- Fentanyl 5 µg/kg, propofol 1 mg / kg.
- Intubation:-Rocuronium 1.2 mg/kg after inj lignocaine 1.5 mg/
- Preinduction B.P was 112/ 82 mmHg & post induction B.P was 96/ 74mm Hg. Ventilation with T.V 10ml/kg & R.R 18/min
- Anesthesia was maintained with vecuronium, Fentanyl, midazolam, isoflurane & O2/N2O mixture.
- The patient was placed in right up or left lateral position and right sided thoracotomy was done.
- During manipulation of tumour, B.P ↑ 182/110 mm Hg & treated with sodium nitroprusside IVI.
- During retraction of lung, SpO2 ↓ to 91% & 100% O2 was given to increase it to 99-100%.
- After the ligation of the vein, B.P ↓ to 60/40 mm Hg & was treated with volume infusion, dopamine, noradrenaline infusion, phenylephrine bolus.
- The tumor was removed.
- The patient received 2000 ml RL, 500 ml hydroxyl ethyl starch and 1000 ml of 5% dextrose during 3 hrs surgery to keep the CVP at around 10 mmHg.
- The 5% dextrose was infused after removal of the tumour to prevent hypoglycemia. Blood glucose was monitored every half an hour during surgery and was within 120-180 mg/dl.
Extubation was done in deep plane of anesthesia with xylocard 3 to 4 ml administered 2 minutes before extubation. Monitoring of the vitals and cardiovascular status postoperatively in the intensive care unit was done which is a mandatory prerequisite.
Conclusion
A lack of controlled studies precludes definitive statements about anesthetic management of patients with pheochromocytoma. It is known that the symptoms of paroxysmal hypertension, sweating, and headache are highly suggestive of the diagnosis. It appears that mortality can be reduced by preoperative α-adrenergic receptor blockade with progressively increasing doses of a blocking agent for 10 days to 2 months for treatment of symptoms, by treatment of myocarditis, and by restoration of intravascular volume. In fact, the largest decrease in mortality of patients after pheochromocytoma resection has occurred with the introduction of preoperative α-adrenergic receptor blockade. The authors believe that knowledge on the part of the anesthetist about the pathophysiology of pheochromocytoma, preoperative patient preparation and communication between surgeon and anesthetist are more important to patient outcome than is the choice of the anesthetic or muscle-relaxing agent.
References
- Golden SH, Robinson KA, Saldanha I, Anton B, Ladenson PW. Clinical review: Prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009; 94: 1853-1878.
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014; 99: 1915-1942.
- Myklejord DJ. Undiagnosed pheochromocytoma: The anesthesiologist nightmare. Clin Med Res 2004; 2: 59-62.
- O’Riordan JA. Pheochromocytomas and anesthesia. Int Anesthesiol Clin. 1997; 35: 99-127.
- Roizen MF, Horrigan RW, Koike M, Eger IE, Mulroy MF, Frazer B, et al. A prospective randomized trial of four anesthetic techniques for resection of pheochromocytoma. Anesthesiology. 1982; 57.
- Iseri LT, Henderson HW, Derr JW. Use of adrenolytic drug, regitine, in pheochromocytoma. Am Heart J 1951; 42: 129-136.
- Mazza A, Armigliato M, Marzola MC, Schiavon L, Montemurro D, Vescovo G, et al. Anti-hypertensive treatment in pheochromocytoma and paraganglioma: Current management and therapeutic features. Endocrine. 2014; 45: 469-478.
- Mihai R, Sadler GP, Bridge H. Adrenergic blockade with phenoxybenzamine and propranolol in a cohort of 60 patients undergoing surgery for phaeochromocytoma. Eur J Anaesthesiol. 2008; 25: 508-510.
- Mannelli M. Management and treatment of pheochromocytomas and paragangliomas. Ann N Y Acad Sci. 2006; 1073: 405-416.
- Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007; 92: 4069-4079.
- Sawka AM, Jaeschke R, Singh RJ, Young WF Jr. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamine’s. J Clin Endocrinol Metab. 2003; 88: 553-558.
- Lenders JW, Pacak K, Walthar MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: Which test is best? JAMA. 2002; 287: 1427-1434.
- Mukherjee JJ, Peppercorn PD, Reznick RH, Patel V, Kaltsas G, Besser M, et al. Pheochromocytoma: Effects of non-ionic contrast medium on circulating catecholamine levels. Radiology. 1996; 202: 227-231.
- Bravo EL. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocrinol Rev. 1994; 15: 356-368.
- Whalen RK, Althausen AF, Daniels GH. Extra-adrenal pheocromocytoma. J Urol. 1992; 147: 1-10.
- Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev. 2003; 24: 539-553.
- Cooper Z, Mihm F. Blood pressure control with fenoldopam during excision of a pheochromocytoma. Anesthesiology. 1999; 41: 558-560.
- Pivalizza EG. Magnesium sulfate and epidural anesthesia in pheochromocytoma and severe coronary artery disease. Anesth Analg. 1995; 81: 414-416.
- Sprung J, O'Hara JF, Gill IS, Abdelmalak B, Sarnaik A, Bravo EL. Anesthetic aspects of laparoscopic and open adrenalectomy for pheochromocytoma. Urology. 2000; 55: 339-343.
- Gill IS. The case for laparoscopic adrenalectomy. J Urol. 2001; 166: 429-436.
Roizen MF, Schreider BD, Hassan SZ. Anesthesia for patients with pheochromocytoma. Anesthesiol Clin North America. 1987; 5: 269-275.
Bogolioubev A, Keefe D, Groeger J. Circulatory shock. Crit Care Clin. 2001; 17: 697.