
Abstract
Human systemic blood system offers larger resistance to the blood flow than pulmonary system: the Left Ventricle (LV) pumps blood more forcefully than the Right Ventricle (RV). The difference in pumping action may arise from the more efficient interaction between actin and myosin in the LV, from the more efficient operation of other sarcomericproteins or from the morphological differences between ventricles. These questions cannot be answered by using whole ventricles or isolated myocytes because the number of molecules involved in tension generation is of the order of 1011. Averaging data from such large number of molecules makes unequivocal characterization of any differences impossible. Measurements must be taken from a few molecules. Moreover, data must be obtained in-situ to account for the molecular crowding effects that are likely to occur in crowded environment such as muscle. We measured kinetics of actomyosin cycle in contracting myocytes from failing ventricles by analyzing fluctuations in orientation of a few actin and myosin molecules in situ. Fluctuations in orientation were caused by ATP-induced repetitive cycles of binding-dissociation of myosin from actin. In both left and right failing ventricles the rate constants characterizing contraction were identical. We also measured distribution of spatial orientations of actin and myosin. The spatial distributions were identical in myocytes from both ventricles.These results show that there is no difference in the way actomyosin interacts with thin filaments in failing left and right ventricles, suggesting that the difference in pumping efficiencies are due either to muscle proteins other than actin and myosin, or that they are due to morphological differences between left and right ventricles.
Keywords: Cross-bridge orientation; Heart ventricles; Fluorescence polarization
Abreviations
ACF: Auto Correlation Function; AP: Alexa633 Phalloidin; EDC: Ethyl-3-[3-(dimethylamino)-propyl]-Carbodiimide; FCS: Fluorescence Correlation Spectroscopy; LV: Left Ventricle; HF: Ventricles from the Failing Heart; NF: Ventricles from Non Failing Heart; OV: Observational Volume; PF: Polarization of Fluorescence; RV: Right Ventricle; SD: Standard Deviation; SSA: Steady State Anisotropy; UP: Unlabeled Phalloidin; XB: Myosin Cross-Bridge
Introduction
The Left Ventricle (LV) has to overcome a large resistance offered by a systemic system, while the Right Ventricle (RV) has to overcome a lesser resistance offered by a pulmonary system. However, it is not clear whether the ability to develop larger force by the LV is due to: 1. More efficient force generation by actin and myosin, 2. More efficient operation of other muscle proteins or 3. Dissimilarities of basic fiber structures of the two ventricles. The left ventricle is composed largely of oblique and circumferential fibers [1], which are known to be more mechanically efficient than the transverse fibers in the free wall of the right ventricle [2,3].
Orientation of the lever arm of myosin head is a defining parameter of force producing interaction between actin and myosin. In order to expose differences in this interaction between left and right ventricles, it is necessary to measure the rate constants governing the interaction, and the spatial distribution of myosin lever arm orientations associated with contraction of each ventricle. Whole ventricles or isolated myocytes cannot be used in such experiments because they contain millions of actomyosin molecules and data originating from so many molecules would get averaged out. All the rate constants of the mechanochemical cycle of actomyosin become unrecoverable, and the final distribution of orientations a large assembly will be a perfect Gaussian, irrespective of whether the data are taken from the left or right ventricle (Central Limit Theorem, [4]). In assessing the actin-myosin interaction, the contribution of individual molecules has to be measured [5,6]. Because of these technical difficulties the question whether individual actin and myosin molecules of LV and RV interact differently has never been asked.
We developed the ability to study few molecules out of millions present in a myocyte. This was possible by focusing on a minute section of a sarcomere. Force-producing interactions between actin and myosin take place the Overlap-band (O-band). We measured kinetics and spatial distribution of a few molecules of actin and myosin in the O-bandof sarcomere inworking failing myocytes. It is important that the measurements be carried in-situ because protein concentration in muscle is high, [7]). Consequently the molecular crowding effects mayplay a role in the operation of muscle [8,9]. We measured fluctuations of orientation of a few actomyosin molecules in situ during contraction. Fluctuations were reported by anisotropy of fluorescence, which is a convenient method to measure conformation changes [10-15]. From fluctuations we calculated the rate constants of myosin interacting with actin.We also compared the spatial distribution of actin and myosin molecules during contraction of both ventricles.
The results show that the kinetics and the steady-state distribution of actin and myosin were the same in contracting myocytes from left and right ventricles from failing human heart It follows that the difference in ventricular function are caused either by non-tension generating muscle proteins or by morphological differences between ventricles.
Materials and Methods
Chemicals and solutions
All chemicals were from Sigma-Aldrich (St Louis, MO) except the fluorescent dye SeTau-647-mono-maleimide which was from SETA BioMedicals (Urbana, IL) and Alexa633 phalloidin (AP) and Unlabeled Phalloidin (UP) were from Molecular Probes (Eugene, OR). The glycerinating solution contained: 50% glycerol, 150 mMKCl, 10mM Tris-HCl pH 7.5, 5 mM MgCl2, 5 mM EGTA, 5 mM ATP, 1 mM DTT, 2 mM PMSF and 0.1% β-mercaptoethanol. Carigor solution contained 50 mMKCl, 10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 0.1 mM CaCl2. Contracting solution contained in addition 5 mM ATP, 20 mMcreatine phosphate and 10 units/ml of 1 mg/ml creatine kinase. EDTA-rigor solution contained 50 mMKCl, 10 mM Tris-HCl pH 7.5, 5 mM EDTA. Glycerinating solution contained 50 mMKCl, 10 mM Tris-HCl pH 7.5, 5 mM MgCl2, 2 mM EGTA, 5 mM ATP, 20 mMcreatine phosphate and 10 units/ml of 1mg/ml creatine kinase.
Preparation of ventricles
Samples of human myocardium were collected at the University of Kentucky using procedures that were approved by the local Institutional Review Board. Failing myocardial samples were procured from patients who received heart transplants. All samples were passed to a researcher as soon as they were removed from the patient and snap-frozen in liquid nitrogen within a few minutes. Samples were shipped to UNTHSC on dry ice. Immediately upon arrival in Fort Worth they placed for 24 hrs in glycerinating solution at 0ºC. After 24 hrs, the glycerinating solution was replaced with a fresh solution and placed at -20ºC. Myocytes (MF) were made from glycerinated hearts after a minimum of 2 weeks at -20ºC.
Preparation of myocytes
2 weeks in a solution containing glycerine and EGTAcaused creation of large holes in the myocyte membrane allowing direct access of MgATP to the O-bands. Myocytes are only ~0.5μm thick thus limiting the Observational Volume to AttoLiters (10-12L, see Figure 2A). Preparation of myocytes involved thorough washing of ventricles with ice-cold EDTA-rigor solution and incubating them for 1hr in this solution in order to wash out ATP present in the glycerinating solution without causing contraction. They were then washed thoroughly with Ca-rigor solutionand homogenized in the Cole-Palmer LabGen 125 homogenizer for 10s followed by further 10s homogenization after a cool down period of 30s. Myocytes were made from ventricles which had spent at least 2 weeks in glycerinating solution at -20ºC, and were used within 2 days of preparation.
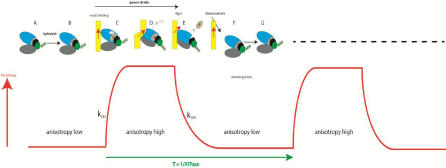
Figure 2: The relation between the anisotropy of actin and the andmechanochemical cycle of actomyosin (drawing of myosin head modified from [Wulf, 2016
#4669]. The cycle begins when myosin is free of actin (yellow) and undergoes hydrolysis [39] (A →B). Upper 50KD domain of myosin head (blue) Lower 50KD
domain (gray) are separate. The lever arm (green coil) is up. Steady state anisotropy (SSA) of F-actin-phalloidin is low (SSA=0.220±0.006). Next, myosin containing
products of hydrolysis binds weakly to thin filaments with a rate constant kON (the red arrow indicates the direction of the dipole moment of the fluorophore attached
to a single actin molecule) (C). Dissociation of Pi ADP and formation of strong binding Actin+Myosin+ADP state (D) is associated with the closing of Upper and
Lower 50KD domains and causes increase of anisotropy of F-actin (SSA=0.248±007). At the same time, transition from weak to strong binding causes power stroke
(indicated by the black arrow). System then undergoes transition to rigor state (D→E), without change of anisotropy. Finally, binding of ATP to the head in rigor
state (E) causes dissociation of myosin with a rate constant kOFF. During dissociation the lever arm (green coil) repositions itself from the orientation pointing down
to the orientation pointing up. The changes in anisotropy of a single Alexa-phalloidin dipole during contraction are shown schematically as a red line.Anisotropy
measurements taken in vitro, with 1μM actin labeled at 10% with phalloidin 488, 0.5μM S1, 2 mM ADP, 5 mM ATP.
Actin labelling
Long-wavelength probe (Alexa633-phalloidin, AP) was selected in order to minimize autofluorescence [16]. 1mg/mL myocytes (approx. 10μM actin) were incubated for 10min in the Ca rigor solution containing in addition 100nM AP+10μM unlabeled phalloidin at room temperature. This procedure assures that only ~1000th actin monomer in a thin filament is fluorescent. Remaining monomers are labeled with non-fluorescent phalloidin. The sparse labeling assures that there only ~16 fluorescent actinsin the OV (see below). After labeling myocytes were spun for 3 min in a desktop centrifuge and gently resuspended in Ca-rigor solution. A 50μL suspension was placed on an ethanol cleaned #1 coverslip coated with polylysine solution to facilitate adhering of myocytes to glass. Myocytes were allowed to adhere to the glass for 10 min before a final wash with Carigor solution by placing drops of solution at one end of a coverslip and sucking out excess solution at the other end with the Whatman #1 filter paper.
Myosin labelling
To measure myosin orientation, Essential Light Chain (ELC) (part of the lever arm of myosin) is labeled with a fluorescent dye. Human ELC contains one SH group, but to increase extinction coefficient another SH group was added by genetic manipulation. Series of steps to include additional thiol group is illustrated in Figure 1.
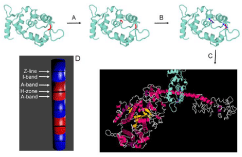
Figure 1: Human ELC containing SH group in position 181 (indicated by red) is genetically produced as described in the text (top left panel). Next, it is genetically
modified to introduce second thiol in position 174 (red in middle top panel, step A). Modified ELC is fluorescently labeled (blue arrows indicate the direction of
transition dipole of the dye, top right panel, step B). Finally, it is exchanged into myocyte myosin in situ (step C). D is a diagram of a myocytes after all steps have
been completed. Fluorescent A-bands are red. Non fluorescent I-bands are blue. Myocytes is cross-linked to prevent any motion. Detail of the procedure are
outlined.
To genetically modify human fast Essential Light Chain (ELC), native LC is subcloned into pQE60 vector to produce pQE-ELC expression vector [17]. The ELC cDNA insert in pQE60-ELC construct contains one Cysat amino acid position 181. We have introduced Cys in place of Gly at amino acid position 174 by site directed mutagenesis to generate pQE60-ELCG174C expression vector. The resulting pQE60-ELCG174C expression vector contained two Cys residues at amino acid positions 174 and 181. The pQE60- ELCG174C construct was generated by PCR-based site directed mutagenesis using the QuickChange kit from Strata Gene (La Jolla, CA), pQE60-ELC template plasmid, and two complimentary primers F-hELCG174C: 5’-gaagtggaagccctgatggcatgtcaaga- agactccaatggctgc-3’ and R-hELCG174C: 5’-gcagccattggagtcttcttgacatgccatcagggcttccacttcttcc-3’. The sequence of the plasmid pQE60-ELCG174C is confirmed by DNA-sequencing of both strands of the entire plasmid. Afterwards, the plasmid pQE60- ELCG174C is introduced into Escherichia coli M15 cells (Qiagen). The expressed recombinant proteins is purified on Ni-NTA-Agarose column and confirmed by immmunobloting with human ELC monoclonal antibody.
Labeling and exchange of ELC into myocytes
SeTau647-maleimide was the dye chosen to label SH groups on ELC’s. It is excited in the far red and hence bypasses most contributions of autofluorescence [16]. SeTau is also resistant to photobleaching (the initial rate = 2.4 s-1) because of nano-encapsulation of the squaraine moiety of the dye chromophore system in a mixed aliphatic-aromatic macrocycle. Very high extinction coefficient (200,000 M-1cm-1), quantum yield (0.65) and large Stokes shift (44nm) makes it ideal for our purpose. Its overall fluorescence intensity was 4.2 times larger than fluorescence intensity of Alexa647. The dye is attached to ELC at positions 174 and 181. 5nM of labeled ELC is exchanged with the native ELC of ventricular myosin in the exchange solution containing 15mM KCl, 5mM EDTA, 5mM DTT, 10mM KH2PO4, 5mM ATP, 1mM Tri-Fluo-Perazine (TFP) and 10mM imidazole, pH 7 [18]. The reaction is allowed to occur at 30°C for ten minutes. The exchange is inefficient because of short exchange time and low temperature. Thus only a small fraction of myosin carry fluorescent label.Since two SeTau molecules per molecule are employed the overall extinction coefficient of the peptide is extremely high (400,000 M-1cm-1). This strategy permits obtaining data from only few molecules of myosin ex-vivo. The dyes are immobilized by myocytes and their combined transition dipoles have a well-defined orientation.
Cross-linking
Failing myocytes contract (shorten) after addition of contracting solution. Shortening would make it impossible to collect data for 20sec from a single spot. Myocytes must be prevented from any movement while preserving the ability to contract (and to develop force). To prevent shortening 1mg/ml myocytes were incubated for 20 min at room temperature with 20mM water-soluble cross-linker 1-Ethyl-3-[3-(Dimethylamino)-propyl]-Carbodiimide (EDC) [19- 26]. The reaction was stopped by adding 20mM DTT. The pH of the solution (7.5) remained unchanged throughout the 20min reaction. As a control we observed cross-linked myocytes under a Nomarski microscope after addition of contracting solution. Myocytes did not shorten or become dehydrated during at least 10min. To check whether cross-linking does not affect ATPase of myocytes, ATPase was measured independently by two researchers (Table 1). The measurements were carried out using Anaspec (Fremont, Ca) Sensolyte MG Phosphate Colorimetric Assay at 30ºC in a 96 well plate and read on a microplate reader at 650nm, with and without crosslinker EDC. Samples were ran in triplicate. Readings were taken for 30 minutes at 2 min. intervals. ATPase of unmodified myocytes was in agreement with [27]. The ATPase was slightly higher for the RV than the LV.
![]()
Ventricle from
Type
Control
Cross-linked
Cross-linked and labeled
6634F LV
Failing
0.040±0.004
0.034±0.004
0.037±0.005
6634F RV
Failing
0.033±0.003
0.037±0.004
0.043±0.005
Table 1: The rates are in number of ATP molecules turned over by 1 myosin molecule per second. 1 mg/mL of myocytes, 5 mMATP, samples ran in triplicate. In every case R2 was 0.98 or 0.99.
Instrument
The data was collected by Pico-Quant MT 200 instrument coupled to the Olympus IX 71 microscope. 10μW laser beam at 635nm modulated at 20 MHz was focused on the O-band by an Olympus x100, NA=1.2 objective. The experimental apparatus was the same as in [28].
Data collection
Before each experiment, the instrument was calibrated to ensure that 100% of emitted light was detected by a parallel or perpendicular channel. Fluorescence of an isotropic solution of a dye with a long fluorescence lifetime (rhodamine 700, excitation at 635nm) was measured. Such dye is practically isotropic because it has no time to reorient itself during ns short laser pulse. Its steady-state anisotropy is 0.400. First the amplifier controlling signal to the perpendicular channel was adjusted to a maximum. Next, the prism was rotated 90oand the amplifier controlling signal to the parallel channel was adjusted to be equal to the signal of parallel channel. Finally, the solution was replaced by a completely anisotropic solution and it was verified that signal reported by each channel was equal. The laser beam was focused by an Olympus x60, NA=1.2 water immersion objective on the O-band of a myocytes. The fluorescence intensity was collected for 20 sec. distributed onto 2,000,000 10μs long bins. To smooth the data, 1000 bins were averaged into 2,000 bins yielding overall time resolution of 10 msec. Fluctuations of Polarization of Fluorescence (PF) and of steady-state anisotropy (r) were computed from the orthogonal components of fluorescent light. The Autocorrelation Function (ACF) was computed from these 2,000 measurements. Huxley at al. determined that a characteristic lifetime of one fluctuation is on the order of milliseconds, so during 20 s the orientation will change X=~20,000 times [29]. The precision of measurement is therefore approximately 1/√X = 0.7%.Approximately 20-30 myocytes were examined in each ventricle.
Estimating the number of observed actin molecules
To determine this number it is necessary to measure fluorescence intensity associated with one molecule of Alexa633-phalloidin. This number was determined by Fluorescence Correlation Spectroscopy (FCS). The autocorrelation function at delay time 0 of fluctuations of freely diffusing AP molecules entering and leaving the OV is equal to the inverse of the number of molecules contributing to the fluctuations N = 1/ACF(0) [30,31]. 15nM Alexa-phalloidin was illuminated with 1-70μW of laser power at 635nm. A calibration curve was constructed by plotting the power of the laser vs. the rate of photon arrival per molecule of the dye. From this curve it was determined that the number of photons/sec collected from a single AP molecule illuminated by a typical laser power used in each experiment (22μW) was ~400 photons/sec/mol. The number of observed actin molecules was estimated as ~16.
Estimating the number of observed myosin molecules
The procedure was identical to the one outlined above for actin, except that 15nM of SeTau dye was used for calibration. A calibration curve showed that that the number of photons/sec collected from a single SeTau molecule was ~900 photons/sec/mol. The number of observed myosin molecules was estimated as ~6.
Time resolved anisotropy measurements
The measurements were performed on FT300 fluorescence lifetime spectrometer (PicoQuant GmbH, Germany). The excitation source was Fianium super-continuum white light source (Fianium Ltd, Whitelase SC400-4). White light was passed through a monochromator to get 635nm light at 10MHz repetition rate. The emission was observed at 665nm through a long pass 650nm filter by fast microchannel photomultiplier tube. The resolution was kept at 4ps per channel, and the pulse width was less than 100 ps. For measuring anisotropy decays, the fluorescence intensity decays were collected while orienting the emission polarizer in vertical and horizontal respective to the vertically oriented excitation polarizer. The vertical (parallel) and horizontal (perpendicular) intensity decays were used to calculate the time dependent anisotropy using the equation:
r(t)=(I_Parallel (t)-I_Perpandicular (t))/(I_Parallel (t)+2I_ Perpandicular (t))
The anisotropy decay was analyzed using Fluofit 4.0 program provided by PicoQuant and was fitted using formula:
r(t)=Σ2592R_i e^(-t/Ø_i )
Where, r is the total anisotropy, Ri is the fractional anisotropy amplitude associated with component, φi is the rotation correlation time and t is the time in nanoseconds. Fluorescence lifetime was measured with the emission polarizer at 54.70.
Statistical analysis
Autocorrelation function was calculated using Origin 2016 (OriginLab Corporation, Northampton, MA) or using R (version 3.3.1). The autocorrelation curve was fitted with a bi-exponential decay model in order to extract the rate constants using Origin 2016 or XPFIT (version 1.2.1) Alango Ltd. XPFIT employs the Inverse Laplace algorithm in order to numerically invert the decay time domain data. The goodness of fit was assessed by chi-squared. Comparisons between groups were performed using an unpaired Student’s t-test by Origin 8.5 program. The differences were deemed significant when P<0.05. Origin 2016 was also used to compute histograms and the autocorrelation functions.
Experimental problems
The difficulty in obtaining single molecule data in-situ is assuring adequate Signal/Noise (S/N) ratio. In-situ data contains significant contributions from autofluorescence and the background due to the dense environment [7]. The background signal consists of a constant fluorescence coming from myosin that is always present in the OV (observing single myosin molecule in-vitro e.g. [32-36] poses no such problem). In contrast to the regular FCS, actin and myosin molecules do not translate and fluorescent signal does not fluctuate between zero and maximum. In our experiments only anisotropy fluctuates around the mean. Moreover, to avoid photobleaching the laser beam cannot be focused on the same spot for much more than 20 sec.
Measuring Orientation of Actin
The rationale of using conformation of F-actin to monitor the kinetics of actomyosin cycle in ventricles is that actin anisotropy reflects the physiological state of a myocyte as illustrated in Figure 2. It shows the relation between the actomyosin cycle and changes of anisotropy of actin.
Head (blue) Lower 50KD domain (gray) are separate. The lever arm (green coil) is up. Steady State Anisotropy (SSA) of F-actin-phalloidin is low (SSA=0.220±0.006). Next, myosin containing products of hydrolysis binds weakly to thin filaments with a rate constant kON (the red arrow indicates the direction of the dipole moment of the fluorophore attached to a single actin molecule) (C). Dissociation of Pi ADP and formation of strong binding Actin+Myosin+ADP state (D) is associated with the closing of Upper and Lower 50KD domains and causes increase of anisotropy of F-actin (SSA=0.248±007. At the same time, transition from weak to strong binding causes power stroke (indicated by the black arrow). System then undergoes transition to rigor state (D→E), without change of anisotropy. Finally, binding of ATP to the head in rigor state (E) causes dissociation of myosin with a rate constant kOFF. During dissociation the lever arm (green coil) repositions itself from the orientation pointing down to the orientation pointing up. The changes in anisotropy of a single Alexa-phalloidin dipole during contraction are shown schematically as a red line.Anisotropy measurements taken in vitro, with 1μM actin labeled at 10% with phalloidin 488, 0.5μM S1, 2 mM ADP, 5 mM ATP.
The main reason for using anisotropy of F-actin to assess the kinetics of actomyosin cycle is that labeling ventricular actin in situ is simple, reproducible and causes no ATPase alteration. Labeling actin withfluorescent phalloidin involves simply gentle irrigation of myocytes, becausephalloidin binds to F-actin extremely strongly. Moreover, the orientation of phalloidin transition dipole reflects orientation of actin [14,15] and is easy to measure [10-15]. Finally, the number of actin molecules that are labeled can be easily controlled [37,38]. The instrument used to measure anisotropy is the same as in [28].
In summary: the ATP-induced interactions of myosin crossbridges with actin result in a rapid momentum transfer from myosin to actin. The force resulting from this transfer is reflected in thealteration of conformation of actin [40,41]. Anisotropy of F-actin is low when it is free or weakly bound to myosin head, or high when it is strongly bound to the myosin head containing ADP or to nucleotide free myosin head (rigor). The cycle repeats itself with the period T (equal to the inverse of ATPase activity).The net result is that anisotropy of phalloidin fluctuates during contraction between low and high values with the rate constants kON and kOFF.
We minimized the number of observed actin molecules by carrying out experiments on the O-band of contracting myocytes isolated from a failing human ventricle. The experimental arrangement is shown in Figure 3.
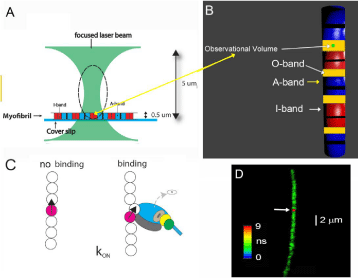
Figure 3: Experimental arrangement. A: A single myocyte is isolated and placed on a cover-slip.Thelaser beam of a confocal microscope is focused to a diffraction
limit (~0.5 μm wide and 4 μm tall, indicated by broken line). The waist of the beam is ~2 μm above a coverslip. The microscope sees only the volume (ellipsoidof-
revolution) outlined by a broken line. Only an I-band (red rectangle) within this volume (called Observational Volume) is fluorescent. Its volume is several Atto
Liters. B: The OV (here shown as a blue-green ellipsoid)is placedin the part of sarcomere where actin and myosin containing filaments overlap) (O-band). Actin
within I-bands is fluorescently labeled (red). The A-bands are non-fluorescent (blue). Overlap bands are orange. Thick black lines are the H-zones; thin black lines
are the Z-bands. Myocytes contracts, but does not shorten because it is cross-linked;C: Myosin bindingor dissociation from actin causes actin to change orientation.
One in one thousand actin molecules within the thin filament is labeled. There are only a few actin molecules within the OV. One fluorescent actin is shown with
the transition dipole moment (black). Neighboring actin molecules may also be changing conformation by the non-fluorescent myosin heads but their orientation
change is not noticed because they are non-fluorescent. Howeverit is possible that myosin head binds that to unlabeled actin monomer close to the labeled one
and this will affect anisotropy [42]. The change may be smaller than when a head binds directly to labeled monomer. The diagram represents the average change
of anisotropy. D: A non-failing human RV myocytesin rigor was imaged by fluorescent lifetime imaging (lifetime confocal image is of better quality than intensity
confocal image). Nevertheless, quality of the image was poor becauseon average only ~16 actin molecules/half-sarcomere were labeled. The color bar indicates
the lifetime (in nanoseconds) of a given pixel. The scale bar is 2 μm. The red dot (pointed by the white arrow) is the 2D projection of the OV. The image has not
been contrast enhanced.1μM actin labeled at 10% with phalloidin 488, 0.5μM S1.
Kinetics of orientation changes of actin
The first parameter that we studied to reveal characteristics of human ventricles was kinetics of orientation changes of actin as illustrated in Figure 3C. Figure 4 highlights again the conformational transitions under study:

Figure 4: Conformational transitions of actin (yellow) during the A: binding of a cross-bridge to a thin filament, characterized by the rate constant kON, B: dissociation
of a cross-bridge from a thin filamentcharacterized by the rate constant kOFF.
Failing ventricular myocytes contracted after addition of a solution containing ATP [28]. They are cross-linked so do not shorten, but retain full ATPase activity. As noted before, changes of orientation of actin during contraction occur because momentum is rapidly transferred from myosin cross-bridges to actin. Red line in Fig. 1 illustrates the changes of anisotropy of a single Alexa-phalloidin molecule during one ATPase cycle of contraction. It is ON-OFF transition in which the ON transition is the rate with which myosin head binds strongly to thin filaments, and OFF transition is the rate of head dissociation from thin filaments.
Experimentally we were able to observe changes in 16 actin molecules (see Materials and Methods). The method introduced by Magde and Elson [31,43,44] was used to extract the rate constants from data: Autocorrelation Function (ACF) of fluctuations of anisotropy is calculated. It is an average of the sum of products of the instantaneous values of PF and the values of PF delayed by delay time τ from 0 τ0 2 sec. An example of ACF is shown in Figure 5.
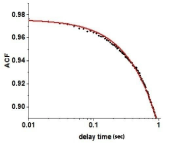
Figure 5: A typical autocorrelation function of fluctuations of actin orientations during contraction of failing right ventricle. Data points are indicated by filled circles.
Red line is a non-linear fit. Number of observed actins was ~16. It should be emphasized that as long as the number of molecules is small, the exact number does
not matter. Observing 16 or 50 molecules would have yielded the same ACF. The fact that the function is not decaying to zero is due to the fact that the background
fluorescence is large.
ACF reveals existence of two different states. The theoretical ACF of the ON-OFF process is shown in Eq. 1 [45].
ACF= ((a_OFF k_ON+ a_ON k_OFF)2)/[(k_OFF+ k_ON)]^2 + k_OFF k_ON [(a_OFF-a_(ON))]^2/[(k_OFF+ k_ON)]^2 e^(- (k_OFF+ k_ON )τ) Eq. 1
wherekON and kOFF are the ON and OFF rate constants and aON, aOFF is the magnitude of anisotropy change (determined by the fitting program). Experimental ACF is fitted to eq. 1 by the nonlinear fit from which the rates kON, and kOFFare obtained. The data is summarized in Table 2.
![]()
Contraction of myocytes from failing heart
kON(s-1)
kOFF (s-1)
LV
0.98±0.24
0.28±0.02
RV
1.37±0.42
0.13±0.11
Table 2: Summary of 153 experiments on RV and 148 experiments on LV from 5 failing hearts. The difference between LV and RV for the rate constant kON was statistically insignificant (P=0.469, t=0.794) and 95 degrees of freedom. The difference between LV and RV for the rate constant kOFF was statistically insignificant (P=0.238, t=1.274) and 90 degrees of freedom. The errors are SEM. The error is larger for kON because there is less experimental points for faster process.
Spatial distribution of actin in contracting failing myocytes
An additional parameter characterizing actin is the distribution of orientations during contraction. The distribution was Gaussian. It was characterized by a Full Width at Half Maximum (FWHM). It indicates the width of the distribution of orientations. Examples of measurements of spatial distribution from 148 experiments from LVs and 153 experiments from RVs are shown in Figure 6. All the data is summarized in Table 3.
![]()
Contraction of ventricles from failing heart
FWHM
AR2
LV
0.348±0.013
0.896±0.018
RV
0.356±0.421
0.888±0.036
Table 3: The average values of distribution of actin orientations in thin filaments during contraction of 148 LVs and 153 RVs myocytes from failing human heart. Errors are SEM. AR² measures goodness of fit (closer to 1 the better).The difference in the mean value of FWHM was statistically insignificant (t = 0.393, P=0.704) with 61 degrees of freedom. The difference in the value of AR² was statistically insignificant (t = 0.388, P=0.707) with 61 degrees of freedom.
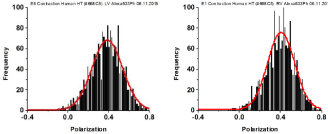
Figure 6: Examples of distribution of actin orientations. Contraction of myocytes from LV (left panel) and RV (right panel). The red line is the best Gaussian fit.
Polarization of Fluorescence (PF) rather than steady-state anisotropy was used here to describe orientation of the lever arm because it has been used routinely to
measure conformation of XBs [12,13,21,46-49]. PF is closely related to the SSFA by SSFA=2PF/(3-PF). PF is the difference between ║ and ┴ components of the
fluorescent light emitted by the dye, normalized by their sum.
Measuring Orientation of Myosin
The direct way to report on conformation changes within ventricular myocytes is to follow changes of orientation myosin. However, following orientation of myosin in-situ is not easy: it involves genetically engineering Essential Light Chain 1 (LC; see Materials and Methods), labeling it with the long-wavelength probe and replacing original LC with fluorescent one without affecting ATPase of a ventricle.The anisotropy of fluorescence reflects conformation changes of fluorescently labeled myosin of skeletal muscle fiber or myocytes [10-13]. The instrument used to measure anisotropy was the same as in [28].
How the anisotropy of labeled LC reflects the kinetics of a myosin head seen in Figure 7.
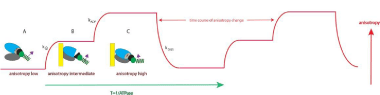
Figure 7: Changes of conformation of a cross-bridge during the contractile cycle. The kinetics of orientation changes of the lever arm is a characteristic property of
muscle. The kinetics are measured by changes of anisotropy of the myosin lever arm which is engineered to contain fluorescent light chain. Light chain orientation
(more precisely orientation of fluorescent dipole moment) is indicated by the magenta arrow; A: XB is originally free of actin. Lower and upper 50KDa domains
of myosincross-bridge (XB) (referred to alternatively as myosin head; gray and blue, respectively) are separated. The lever arm is facing down). Steady State
Fluorescence Anisotropy (SSFA) of the lever arm is low. B: The anisotropy of LC+myocyte in rigor complex. Lower and upper 50KDa domains of XB are closed.
The anisotropy assumes intermediate value; C: Myosin head containing ADP is bound to actin. Lower and upper 50KDa domainsare closed, the anisotropy of a
complex assumes the highest value. This is consistent with recent report [50]. 0.1 mg/mL myocytes from the left ventricle, 2 mMMgADP, 5 mMMgATP. Anisotropy
in-vitro was measured with excitation at 630 nm by Fianiumsupercontinuum white light source. The emission was observed at 665 nm through long pass 650 nm
filter by a fast microchannel photomultiplier tube.
In order to avoid problems associated with averaging data from numerous molecules,just like in the case of actin, the number of observed myosin molecules needs to be minimized. It was done by measuring orientation of myosin in the A-band of contracting myocytes isolated from a human ventricle. The interaction between actin and myosin occur only in the part of an A-band where thick and thin filament overlap (O-band). An A-band contains ~104 myosin molecules, still too many to obtain kinetic information. To reduce this number, myosin was intentionally labeled inefficiently with fluorescent probe. Only a single myosin molecule out of 1000 was labeled. Myocytes were cross-linked with zero-length cross-linker to eliminate any movement during contraction while preserving ATPase activity, Figure 8 explains how the experiments were carried out.
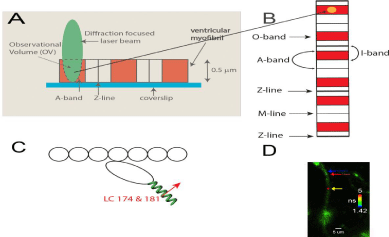
Figure 8: A: A diffraction limited laser beam (green ellipsoid) is focused on an isolated myocytes sitting on a coverslip. A confocal microscope sees only the
Observational Volume (OV, outlined by a broken line). A-band is colored red. B: the interaction between actin and myosin occurs only where actin and myosin
containing filaments overlap at the edges of the A-band (red). Fluorescence is collected from the OV shown hereas anorange sphere projected into theoverlap
band (O-band).The diameter of the OV projectedon the mage plane is equal to the diameter of the confocal pinhole (50 μm) divided by the magnification of the
objective (100x). Myosin within the O-band is fluorescently labeled (red). The I-bands are non-non-fluorescent (white). Thin black lines are the H-zones; thick black
lines are the Z-bands. The myocytes contracts (i.e. develops normal force), but does not shorten because it is cross-linked (see Methods); C: One in approximately
one thousand myosin molecules within the thick filament is labeled at LC Cys 174 and Cys 181 located on the lever arm of myosin (green coil. The transition dipole
moments the dye of two labeled myosins are marked in red (emission dipoles of the dye attached to these two cysteines point in the same direction)n. Labeling of
only 1 in a 1000 myosins ensures that there are only few myosin molecules in the OV (see Methods). Actin monomers (white) are non-fluorescent. D: Fluorescent
lifetime image of a myocytes from a non-failing human RV in rigor. Myocytes were imaged by fluorescent lifetime imaging because a lifetime image is of better
quality than a confocal image. Nevertheless, the quality of the image is poor because of intentionally inefficient labeling. On the average only ~6 myosin molecules/
half-sarcomere were labeled with the fluorophore (see Methods). The color bar indicates lifetime (in nanoseconds) of a given pixel. The non-fluorescent part
(H-zone) is pointed to by the blue arrow. The data was collected only from the part of a myocytes which was aligned vertically (thus it was not collected from the
A-band pointed to by the red arrow), but it was collected from the red spot (pointed to by the yellow arrow.The scale bar is 5 μm. The image has not been contrast
enhanced.
Kinetics of myosin orientation changes
Myocytes were obtained from failing hearts. They do not shorten, but retain full ATPase activity: ATPases of control and cross-linked LV myocytes were 0.040±0.004 s-1/mol; 0.034±0.004 s-1/mol for respectively. ATPases of control and cross-linked RV myocytes were and 0.033±0.003 s-1/mol; 0.037±0.004 s-1/mol respectively.The experiment begins by placing isolated myocytes on an ethanol cleaned cover slip. The conformational transitions are shown as a Figure 9. The cycle begins when XB is dissociated from a thin filament where anisotropy is low. Binding to a thin filament causes a small increase in anisotropy(transition A). Dissociation of Pi and assumption of actinmyosin- ADP complex (transition from apoenzyme to holoenzyme (transition B) causes further increase in anisotropy in accordance with [51]. Finally, dissociation of myosin from actin (transition C) occurs with the rate kDISS.
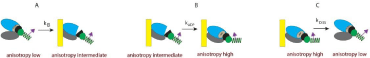
Figure 9: Conformational transitions of XBs during contraction of a ventricle. A: binding of cross-bridges to a thin filament of a ventricle is characterized by the rate
constant kB; B: transition from apoenzyme to holoenzyme form of myosin ischaracterized by the rate constant kADP; C: dissociation of a cross-bridge from a thin
filamentcharacterized by the rate constant kDISS.
The repetitive changes in anisotropy cause intensity fluctuations. The ACF of fluctuations is calculated by the method first introduced by Magde and Elson [31,43,44]. The rate constants are computed from ACF by non-linear fit. The analytical form of ACF of the 3-state process is very complex and is fully described in [45].
Examples of the ACF are shown in [28]. Like ACF shown in Figure 5, the ACF reflects the two state process, even though anisotropy changes occur in three steps.XB binding to actin (rate kB) is too fast to be reflected in the ACF. This is due to the limited time resolution of the instrument. It collects photons every 10μsec, but in order to decrease the noise, the 2 M data points are packed into 2,000 bins, 1,000 points per bin. Thus the instrumental response time is 10 msec. We cannot detect processes faster than 100 Hz (s-1). The rate constants were calculated from ACF’s by non-linear fit to analytical solution, described in [45].
We analyzed myocytes from 5heart failure ventricles. Table 4 shows averages from 5 ventricles. There was no statistical difference between LV and RV for either rate constant.
![]()
HF Contraction
kADP
kDISS
HF LV
1.69±0.05
2.24±0.06
HF RV
1.72±0.04
2.36±0.20
Table 4: Differences between the kinetic rate constants of LV and RV of heart failure ventricles. The results are averages of 27 experiments on LVs and 26 experiments on RVs. Errors are SD. Overall, the differences in the values of HF LV and RV were not significant.
Spatial distribution of the lever arm in contracting failing myocytes
The distribution of orientations of lever arms was Gaussian. Examples of measurements of 27 experiments from LVs and 26 experiments from RVs are shown in Figure 10.
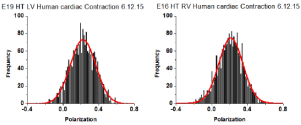
Figure 10: Spatial distribution of lever arm angles of cross-bridges of contracting HF ventricles. The orientation of ~6 cross-bridges in 27 different preparations
of LV and 26 preparations of RV As in the case of actin, the orientation expressed as polarization of fluorescence. Data shows no difference in FWHM. Bars are
data. Red line is the best fit.
All data is summarized in Table 5. The distributions show no differences in the value of FWHM. AR² values indicate how well the fitted curve (red) matches a perfect Gaussian (a perfect fit has an AR² value of 1).
![]()
Contraction
FWHM
AR2
HF LV
0.390±0.03
0.853± 0.096
HF RV
0.381± 0.01
0.889±0.078
Table 5: The FWHM of distribution of lever arm angles of cross-bridges in the O-band of contracting LVs and RVs myocytes from failing human heart. The results are averages of 27 experiments on LVs and 26 experimentson RVs. Errors are SD. By conventional criteria, the statistical significance of difference of FWHM was not statistically significant (t = 0.691, P=0.505) within 10 degrees of freedom. AR²measures goodness of fit (closer to 1 the better).
Discussion
The main conclusion of this paper is the kinetic rates of conformational transitions and the distribution of myosin and actin were the same in contracting in-situactin and myosin from myocytes from failing LV and RV. Consistent with our data, earlier work showed failing RV and LV myocytes displayed similar decrease in development of maximal force [52]. However, this work also revealed interventricular differences in myocyte function in experimental congestive heart failure of rats. LV myocytes were less Ca2+-sensitive than RV myocytes. There were also differences in expression and activation of PKC-α and in phosphorylation of cTnI and cTnT. We did not measure either activation of PKC-α or phosphorylation of cTnI and cTnT, possibly better indicators of myocytes function. Other reasons for the difference between this work and ref [52] is that different species were examined and it is possible that the difference arose because we examined individual molecules while Belin et al. looked at whole myocytes. Individual molecules approach allowsdata collection under in situ conditions thus taking into account molecular crowding and packing of myosin in thick filaments, which may play an important role in crowded systems such as muscle.
Our results are consistent with the fact that actin is expressed from the same genes [53] in both ventricles and that there is no evidence of differential expression of β-myosin heavy chain in ventricles, although ventricular-specific expression was seen during chamber specification in the zebrafish embryo [54].
We were able to examine molecular function of individual molecules because we worked on mesoscopic - not macroscopic - samples. Observing individual molecules in-situ is technically challenging because myosin and actin are always present in the OV. They do not translate but remain fixed in the OV. Unlike in the regular Fluorescence Correlation Spectroscopy (FCS), the fluorescent signal does not fluctuate between zero and maximum; instead it fluctuates around non-zero mean. A further complication is that, to avoid photobleaching, the laser beam cannot be focused on the same spot for much more than 20 sec to. It must also be emphasized that the orientation of myosin the lever arm was measured using recombinant light chain exchanged with endogenous LC. This leaves open the possibility that the data may not reflect exactly on the interactions of endogenous LC which carries no fluorophores. Post translational modifications are unlikely to affect the results because in the mutated clone G has been shown to have Cys at amino acid position 147 by DNA sequencing. Therefore, expression of this mutated plasmid is expected to produce G147C isoform of the protein. The most obvious post translational modification, formation of Cys-Cys link between Cys 147 and 181, is impossible because sample was pre-washed with DTT (ll. 265). The isoform shift will occur upon substituting Gly for Cys and the overall negative charge will decrease.
The data from failing ventricles should be supplemented by data from non-failing ventricles. In view of the fact that human nonfailing ventricles are difficult to obtain, we propose that large animal model of heart failure should be exploited to examine the differences between healthy and diseased ventricles.
Conclusions
• We looked for the differences between kinetics of orientation changes and spatial distributions of actin molecules in actomyosin complex during contraction of myocytes from failing LV and RV.
• We looked for the differences between kineticsof orientation changes and spatial distributionof lever arms of myosin during contraction of myocytes from failing LV and RV.
• Experiments were done in-situ thus accounting for molecular crowding.
• Actin kinetics was identical in contracting myocytes from failing LV and RV.
• Actin molecules in contracting myocytes from failing LV and RV were identically distributed in space.
• Myosin lever arm kinetics was identical in contracting myocytes from failing LV and RV.
• Myosin lever arms in contracting myocytes from LV and RV were identically distributed in space.
• We conclude that the difference in pumping efficiencies are due either to muscle proteins other than actin and myosin, or that they are due to morphological differences between left and right ventricles.
References
- Schwarz K, Singh S, Dawson D, Frenneaux MP. Right ventricular function in left ventricular disease: pathophysiology and implications. Heart Lung Circ. 2013; 22: 507-511.
- Austin EH 3rd. The ventricular myocardial band of Torrent-Guasp-the controversy: an editorial. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2007; 87-88.
- Sallin EA. Fiber orientation and ejection fraction in the human left ventricle. Biophys J. 1969; 9: 954-964.
- Bracewell R, The Fourier Transform and Its Applications. 1965, New York: McGraw-Hill.
- Elson EL. Quick tour of fluorescence correlation spectroscopy from its inception. J Biomed Opt. 2004; 9: 857-864.
- Elson EL. Fluorescence Correlation Spectroscopy: Past, Present, Future. Biophys J. 2011; 101: 2855-2870.
- Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem. 2001; 276: 10577-10580.
- Mourao MA, Hakim JB, Schnell S. Connecting the dots: the effects of macromolecular crowding on cell physiology. Biophys J. 2014; 107: 2761- 2766.
- Aronson JF, Morales MF. Polarization of tryptophan fluorescence in muscle. Biochemistry. 1969; 8: 4517-4522.
- Borejdo J, Putnam S, Morales MF. Fluctuations in polarized fluorescence: evidence that muscle cross bridges rotate repetitively during contraction. Proc Natl Acad Sci USA. 1979; 76: 6346-6350.
- Dos Remedios CG, Millikan RG, Morales MF. Polarization of tryptophan fluorescence from single striated muscle fibers. A molecular probe of contractile state. J Gen Physiol. 1972; 59: 103-120.
- Morales MF. Calculation of the polarized fluorescence from a labeled muscle fiber. Proc Nat Acad Sci USA. 1984; 81: 145-149.
- Borovikov YS, Kuleva NV, Khoroshev MI. Polarization microfluorimetry study of interaction between myosin head and F-actin in muscle fibers. Gen Physiol Biophys. 1991; 10: 441-459.
- Borovikov YS. Chernogriadskaia NA. Studies on conformational changes in F-actin of glycerinated muscle fibers during relaxation by means of polarized ultraviolet fluorescence microscopy. Microsc Acta. 1979; 81: 383-392.
- Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer. 2006.
- Duggal D, Nagwekar J, Rich R, Huang W, Midde K, Fudala R, et al. Effect of a Myosin Regulatory Light Chain mutation K104E on Actin-Myosin Interactions. Am J Physiol Heart Circ Physiol. 2015; 308: 248-2 57.
- Sweeney HL. Function of the N-terminus of the myosin essential light chain of vertebrate striated muscle. Biophys J. 1995; 68: 112-119.
- Corrie JE, Brandmeier BD, Ferguson RE, Trentham DR, Kendrick-Jones J, Hopkins SC, et al. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nature. 1999; 400: 425-430.
- Corrie JE, Craik JS, Munasinghe VR. A homobifunctional rhodamine for labeling proteins with defined orientations of a fluorophore. Bioconjug Chem. 1998; 9: 160-167.
- Hopkins SC, Sabido-David C, van der Heide UA, Ferguson RE, Brandmeier BD, Dale RE, et al. Orientation changes of the myosin light chain domain during filament sliding in active and rigor muscle. J Mol Biol. 2002; 318: 1275- 1291.
- Lewis JH, Beausang JF, Sweeney HL, Goldman YE. The azimuthal path of myosin V and its dependence on lever-arm length. J Gen Physiol. 2012; 139: 101-120.
- Herrmann C, Lionne C, Travers F, Barman T. Correlation of ActoS1, myofibrillar, and muscle fiber ATPases. Biochemistry. 1994; 33: 4148-4154.
- Tsaturyan AK, Bershitsky SY, Burns R, Ferenczi MA. Structural changes in the actin-myosin cross-bridges associated with force generation induced by temperature jump in permeabilized frog muscle fibers. Biophys J. 1999; 77: 354-372.
- Bershitsky SY, Tsaturyan AK, Bershitskaya ON, Mashanov GI, Brown P, Burns R, et al. Muscle force is generated by myosin heads stereospecifically attached to actin. Nature. 1997; 388: 186-190.
- Barman T, Brune M, Lionne C, Piroddi N, Poggesi C, Stehle R, et al. ATPase and shortening rates in frog fast skeletal myofibrils by time-resolved measurements of protein-bound and free Pi. Biophys J. 1998; 74: 3120-3130.
- Alpert NR, Gordon MS. Myofibrillar adenosine triphosphatase activity in congestive heart failure. Am J Physiol. 1962; 202: 940-946.
- Duggal D, Requena S, Nagwekar J, Raut S, Rich R, Das H, et al. No Difference in Myosin Kinetics and Spatial Distribution of the Lever Arm in the Left and Right Ventricles of Human Hearts. Frontiers. in press. 2017.
- Huxley AF, Simmons RM. Proposed mechanism of force generation in striated muscle. Nature. 1971; 233: 533-538.
- Elson EL, Magde D. Fluorescence Correlation Spectroscopy: Coceptual Basis and Theory. Biopolymers. 1974; 13: 1-28.
- Magde D, EL Elson, Webb WW. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 1974; 13: 29-61.
- Forkey JN, Quinlan ME, Shaw MA, Corrie JE, Goldman YE. Threedimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 2003; 422: 399-404.
- Forkey JN, Quinlan ME, Goldman YE. Measurement of single macromolecule orientation by total internal reflection fluorescence polarization microscopy. Biophys J. 2005; 89: 1261-1271.
- Walcott S, Warshaw DM, Debold EP. Mechanical Coupling between Myosin Molecules Causes Differences between Ensemble and Single-Molecule Measurements. Biophys J. 2012; 103: 501-510.
- Baker JE, Brosseau C, Joel PB, Warshaw DM. The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophys J. 2002; 82: 2134–2147.
- Pate E, Cooke R. Simulation of stochastic processes in motile crossbridge systems. J Muscle Res Cell Motil. 1991; 12: 376-393.
- Borejdo J, Shepard A, Dumka D, Akopova I, et al. Changes in Orientation of Actin during Contraction of Muscle. Biophys J. 2003; 86: 2308-2317.
- Borejdo J, Shepard A, Dumka D, Akopova I, Talent J, Malka A, Burghardt TP. Changes in orientation of actin during contraction of muscle. Biophys J. 2004; 86: 2308-2317.
- Lymn RW, Taylor EW. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry. 1971; 10: 4617-4624.
- Yanagida T, Oosawa F. Polarized fluorescence from epsilon-ADP incorporated into F-actin in a myosin-free single fiber: conformation of F-actin and changes induced in it by heavy meromyosin. J Mol Biol. 1978; 126: 507- 524.
- Yanagida T, Oosawa F. Conformational changes of F-actin-epsilon-ADP in thin filaments in myosin-free muscle fibers induced by Ca2+. J Mol Biol. 1980; 140: 313-320.
- Ando T. Propagation of Acto-S-1 ATPase reaction-coupled conformational change in actin along the filament. J Biochem (Tokyo). 1989; 105: 818-822.
- Elson EL. Fluorescence correlataion spectroscopy and photobleaching recovery. Annu Rev Phys Chem. 1985; 36: 379-406.
- Elson EL. Introduction to FCS. Short Course on Cellular and Molecular Fluorescence. Gryczynski Z, editor. Fort Worth: UNT. 2007; 2: 1-10.
- Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, Zhao J, et al. Cross-bridge Kinetics in Myofibrils Containing Familial Hypertrophic Cardiomyopathy R58Q Mutation in the Regulatory Light Chain of Myosin. J Theor Biol. 2011; 284: 71-81.
- Nihei T, Mendelson RA, Botts J. Use of fluorescence polarization to observe changes in attitude of S1 moieties in muscle fibers. Biophys J. 1974; 14: 236-242.
- Tregear RT, Mendelson RA. Polarization from a helix of fluorophores and its relation to that obtained from muscle. Biophys J. 1975; 15: 455-467.
- Sabido-David C, Brandmeier B, Craik JS, Corrie JE, Trentham DR, Irving M. Steady-state fluorescence polarization studies of the orientation of myosin regulatory light chains in single skeletal muscle fibers using pure isomers of iodoacetamidotetramethylrhodamine. Biophys J. 1998; 74: 3083-3092.
- Hopkins SC, Sabido-David C, Corrie JE, Irving M, Goldman YE. Fluorescence polarization transients from rhodamine isomers on the myosin regulatory light chain in skeletal muscle fibers. Biophys J. 1998; 74: 3093-3110.
- Wulf SF, Ropars V, Fujita-Becker S, Oster M, Hofhaus G, Trabuco LG, et al. Force-producing ADP state of myosin bound to actin. Proc Natl Acad Sci USA. 2016; 113: E1844-E1852.
- Coureux PD, Sweeney HL, Houdusse A. Three myosin V structures delineate essential features of chemo-mechanical transduction. Embo J. 2004; 23: 4527-4537.
- Belin R, Sumandea MP, Seivert GA, Harvey LA, Geenen DL, Solaro RJ. Interventricular differences in myofilament function in experimental congestive heart failure. Pflugers Arch. 2011; 462: 795-809.
- Wessels MW, Willems PJ. Mutations in sarcomeric protein genes not only lead to cardiomyopathy but also to congenital cardiovascular malformations. Clin Genet. 2008; 74: 16-19.
- England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. 2013; 70: 1221-1239.
- Wulf SF, Ropars V, Fujita-Becker S, Oster M, Hofhaus G, Trabuco LG, et al. Force-producing ADP state of myosin bound to actin. Proc Natl Acad Sci USA. 2016; 113: E1844-E1852.