
Abstract
While anthracyclines are a class of chemotherapeutic agents that have improved the treatment of a broad spectrum of malignancies, it is one of the most cardiotoxic agents used to treat the tumor. Despite their dose-dependent cardiotoxicity, anthracyclines-based chemotherapy has become the main stay of tumor therapy due to its efficacy. Meanwhile, Statins, widely used in the clinical setting to reduce serum cholesterol levels and to reduce cardiac morbidity and mortality, have pleiotropic biological effects independent of their lipid-lowering effects. Many studies at home and abroad believe that statins can prevent the cardiotoxicity caused by anthracyclines and propose that statins can play beneficial pleiotropic cardiovascular effects through anti-inflammatory and antioxidant mechanisms, thus playing a cardioprotective effect. Illuminating the mechanisms of Anthracyclines-induced cardiotoxicity and Statins associated therapeutic continue to be the main focus in the field of anti-tumor. Therefore, we summarized the current research surrounding the mechanisms of anthracyclines-induced cardiotoxicity and the protective effect of statins on anthracyclines-induced cardiotoxicity and its possible mechanism.
Keywords: Statins; Anthracyclines; Cardiotoxicity; Mechanism; Anti-tumor
Introduction
Anthracyclines such as Doxorubicin (DOX), daunorubicin, idarubicin and so on are a family of potent chemotherapeutics that are broadly used to treat solid tumors (ovary, breast, stomach, brain, and gastrointestinal tumors) and hematological malignancies (lymphoma and pediatric leukemia) [1,2], Anthracyclines can be used alone or combined with other antitumor regimens, such as radiation therapy or monoclonal antibodies [3,4]. However, cardiotoxicity arise as a leading cause of morbidity and mortality among tumor survivors receiving anthracyclines therapy, largely limiting the clinical application of these drugs [5]. Anthracyclines-induced cardiotoxicity involves direct effects of the chemotherapeutics on cardiovascular function and structure, or may be due to accelerated development of cardiovascular diseases. The risk of cardiomyopathy is more than 30% at a cumulative dose of DOX of 400mg/m² and more than 50% at a cumulative dose of 500mg/m² [6]. Even at lower cumulative doses of 240mg/m2, cardiomyopathy can occur in 10–15% of patients and clinical HF (HF) in more than 5% [6]. Anthracyclines-induced cardiotoxicity proves an unresolved major problem in related antitumor therapy, which side effect of inducing cardiac dysfunction has hampered their clinical use. And the mechanism underlying anthracyclines cardiotoxicityremains obscure, increasing evidence points to Anthracyclines-induced Reactive Oxygen Species (ROS), mitochondria damage, Ca2+ overload and iron ferroptosis, cell autophagy and disruption of cardiac metabolism as major targets of anthracyclines--induced cardiotoxicity.
Anthracyclines-Induced Cardiotoxicity
Clinical manifestations of anthracyclines-induced cardiotoxicity: Anthracyclines-induced cardiotoxicity was unrecognized until 1967 when Karnofsky first observed that anthracyclines were associated with chronic Heart Failure (HF) [7]. According to the time of onset, anthracyclines-induced cardiotoxicity presents acutely, early and lately. Acute cardiotoxicity, predominantly supraventricular arrhythmia, transient LV dysfunction and Electrocardiographic (ECG) changes, develops in <1% of patients instantly after infusion and proves usually reversible. However, acute cardiac dysfunction may also evolve into early or late cardiotoxicity [5]. Three distinct types of Anthracyclines-induced cardiotoxicities have been recognized: 1) “acute”, occurring after a single dose or course, within two weeks from the end of treatment; 2) “early onset chronic”, developing within 1 year, is the most frequent and clinically relevant form of cardiotoxicity, usually presenting as a dilated and hypokinetic cardiomyopathy leading to HF; 3) “late onset chronic” developing years, or even decades, after the end of chemotherapy [8]. In the Childhood Cancer Survivor Study, a study of 14358 5-year survivors of childhood malignancies, use of <250mg/m² of anthracyclines were associated with a 2.4-fold higher risk of developing congestive HF compared to those patients who did not receive anthracyclines. This risk increased to 5.2-fold with the use of ≥250mg/m² of DOX [9]. Patients treated with commonly used anthracyclines doses and >65 years old, the rate of Anthracyclines-induced HF even be as high as 10% [10]. Cardinale conducted a prospective study involving 2625 patients with a mean follow-up of 5.2 years that demonstrated a 9% overall incidence of cardiotoxicity with anthracyclines treatment and 98% of patients occurred within the first year and were asymptomatic [11]. In a retrospective study of 640 patients on DOX, which defined cardiotoxicity as Left Ventricular Ejection Fraction (LVEF) <50% with a decrease in >10 absolute points. 32 patients (5%) developed chronic HF. Of those, 38% had mild HF (New York Heart Association (NYHA) Class I or II), 34% developed moderate HF (NYHA Class III) and 28% experienced severe HF (NYHA Class IV) [10]. It is now well established that anthracyclines induced-cardiotoxicity is dose dependent. One large study demonstrated that left ventricular dysfunction (defined as reduction in ejection fraction of >10% below normal) occurred in 10%, 16%, 32%, and 65% at cumulative DOX doses of 250, 300, 400, and 550mg/m² respectively [12]. Thus, even at the lowest dose, anthracyclines can also result in significant left ventricular dysfunction. Risk factors for Anthracyclines-induced cardiotoxicity include lifetime cumulative dose, infusion regimen and any condition that increases cardiac susceptibility, such as pre-existing cardiovascular disease, hypertension, concomitant use of other chemotherapies or mediastinal radiation therapy and older age (>65 years) [13]. Therefore, early detection of cardiac dysfunction is crucial to prevent anthracyclines-induced cardiotoxicity. If anthracyclines-induced cardiac dysfunction is identified early and interfered with HF medications, patients recover easily. In contrast, HF is difficult to treat if detected late after anthracyclines therapy [14].
Mechanisms of anthracyclines-induced cardiotoxicity: The mechanism of anthracyclines-induced cardiotoxicity is not yet clearly. There exist many hypotheses about the mechanism of anthracyclines-induced cardiotoxicity, including oxidative stress hypothesis, cell energy metabolism theory, Ca2+ overload theory, apoptosis theory, immune response theory and so on. The two main accepted hypotheses are as follows and not mutually exclusive: (1) Oxidative stress, which in the presence of iron, generates reactive oxygen species that cause lipid peroxidation of the cell membrane leading to damage of the cardiomyocytes [1,15,16]. (2) Inhibition of topoisomerase 2β, which is active in quiescent non-proliferating cardiomyocytes, can result in the activation of cell death pathways and inhibition of mitochondrial biogenesis [15,16].
Oxidative Stress Hypothesis
Studies showed that oxidative and nitrifying stresses are the key to the anthracyclines induced-cardiotoxicity, which can lead to the oxidation of macromolecules such as lipids, nucleic acids and proteins and disrupt cell function. Anthracyclines can form semiquinone free radicals by losing electrons, and the lost electrons always combine with oxygen to form superoxide radicals, which disproportionately or spontaneously form H2O2. Under the catalysis of superoxide dismutase, H2O2 and superoxide radicals can produce the hydroxyl radicals (OH-), more reactive and toxic. Semiquinone radicals can mediate ROS increase by inhibiting the oxidative cycle of respiratory chain complex I on the mitochondrial inner membrane and gain electrons from NADH or NADPH to form anthracyclines again. Under iron catalysis, a small dose of anthracyclines rapidly generates a large amount of ROS through this cycle to promote lipid peroxidation, resulting in more toxic and stable aldehydes. These aldehydes diffuse and destroy intracellular macromolecules easily [17,18]. Lipid peroxidation usually initiates the arachidonic acid pathway through activation of phospholipase A2 to induce inflammation and apoptosis in vascular endothelial cells. For cardiomyocytes lacking antioxidant enzymes, their antioxidant activity is weaker than other tissue cells. Anthracyclines can significantly reduce the levels of antioxidant enzymes such as superoxide dismutase and catalase, increase the accumulation of reactive oxygen species and further aggravate oxidative stress. Most of the negatively charged cardiolipin exists in the mitochondrial inner membrane and myocardium, which is easy to combine with positively charged anthracyclines to form a stable complex leading to the uncoupling of the respiratory chain and affecting the process of oxidative phosphorylation [1,19]. Cardiomyoblast treated with DOX shows an increased level of NADPH oxidase (NOX) and flavin-containing enzymes such as P450 reductase, nitric oxide synthase leading to increased level of reactive oxygen species and subsequently oxidative stress [20]. In addition to reactive oxygen species, reactive nitrogen as well as plays a key role in the cardiotoxicity caused by DOX. The mechanism may be an increased expression of cardiomyocyte induced nitric oxide synthase and the superoxide ions formed in the DOX redox cycle quickly combine with nitric oxide to produce a powerful oxidant peroxynitrite, which further activates matrix metalloproteinase, causing extracellular matrix remodeling, fibroblast proliferation and collagen deposition, leading to myocardial tissue damage [21]. In addition, anthracyclines can not only increase intracellular iron by activating iron regulatory protein, but also chelate iron ions and trigger the generation of oxygen free radicals, especially hydroxyl free radicals, causing lipid peroxidation of cardiomyocyte membrane and damage of myocardial mitochondrial DNA damage [22].
Inhibition of Topoisomerase 2β
Human DNA topoisomerases are classified into two classes based on structure and mechanisms. Topoisomerase 1 catalyze the formation of DNA single-strand breaks during the catalytic cycle, whereas Topoisomerase2 (Top2) introduce Double-Strand Breaks (DSBs) in the DNA template [23]. The target of anthracyclines action is Top2 which has two isoform, Top 2α and Top 2β. The former exists in rapidly dividing cells such as cancer cells and can combine with anthracyclines to form a lytic complex inducing tumor cell apoptosis. It is considered to be the molecular basis of anti tumor activity. The latter mainly exists in human cardiomyocytes and combines with anthracyclines to form a cleavage complex [24]. On the one hand, this cleavage complex can lead to DNA damage, protein synthesis disorder and specific gene transcription barrier in cardiomyocytes. On the other hand, it presents a strong apoptotic stimulus to activate the P53 apoptosis signaling pathway-dependent mitochondrial dysfunction. It affects oxidative phosphorylation and mitochondrial biosynthesis in cardiomyocytes, causing cardiomyocyte apoptosis and leads to HF [25]. In a mouse model of cardiomyocyte-specific deletion of Top2β gene, the lack of Top2β in heart cells was shown to protect mice from DOX-induced heart cell damage and development of progressive HF [15]. Similar genotoxic mechanisms as well as occur in mitochondrial DNA [17]. Anthracyclines can induce mitochondrial damage because of uncoupling of the electron transport chain, disruption of mitochondrial membrane potential and production of ROS, especially in combination with the disruption of mitochondrial iron metabolism [26]. Cardiomyocytes are rich in mitochondria, which further aggravates anthracyclines-induced cardiotoxicity. Indeed, animal studies demonstrated that chronic Anthracyclines-induced HF relates with imbalances in mitochondrial mass and reduced expression of genes regulating mitochondrial homeostasis [27,28]. But whether TOP2-dependent DSBs in mtDNA or TOP2-independent oxidative mtDNA base lesions cause depletion of mtDNA content is hard to discriminate [29].
Ca2+ Overload and Iron Metabolism Disorder
In normal cardiomyocytes, Ca2+ mostly exists in the sarcoplasmic reticulum, mitochondria and sarcolemma, and plays an important role in the excitation-contraction coupling of cardiomyocytes. Anthracyclines-induced cardiotoxicity is associated with the dysregulation of Ca2+ level. The impaired Ca2+ homeostasis acts as a reason for reactive oxygen species generation [20]. Anthracyclines can act on cardiomyocytes, activate the Ca2+ channels in the sarcoplasmic reticulum and increase the amount of Ca2+ released from the sarcoplasmic reticulum to the cytoplasm [30]. The increase of intracellular free Ca2+ concentration can affect the electrical activity of cardiomyocytes, leading to various types of arrhythmias. Anthracyclines can also inhibit the gene expression of Ca2+-ATPase on the sarcoplasmic reticulum of cardiomyocytes, there by affecting the biosynthesis of Ca2+-ATPase, reducing its activity, reducing the ability of the sarcoplasmic reticulum to absorb Ca2+ and reducing the production of ATP by mitochondria, myocardial energy metabolism disorders, aggravate cell damage, and even lead to myocardial cell death [31]. Ca2+ can increase the expression of NF-kB by activating p38MAP kinase and JNK pathway and activate pro-apoptotic genes such as BAX and BAD, resulting in causing apoptosis and inflammation [32] (Figure 1).
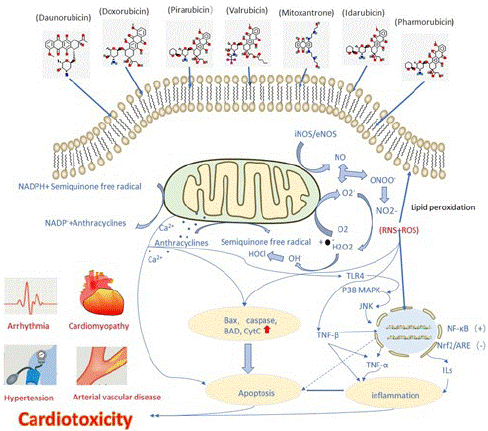
Figure 1: Schematic diagram showing the generation of anthracyclines-induced oxidative stress and nitrative stress. Oxygen free radicals combine with NO to generate peroxynitrite. Ca2+ overload mediates apoptosis via BAX/Caspases and also initiates the TLR-4 mediated activation of p38 and JNK. Calcium accumulation leads to the activation of NF-kB that further increases the expression of pro-inflammatory cytokines (TNF-α and ILs). Nitrative and oxidative stress collectively activates TGFβ which suppresses Nrf2/ARE signaling pathway. The oxidative and nitrative stress-mediated apoptosis and inflammation eventually lead to cardiotoxicity.
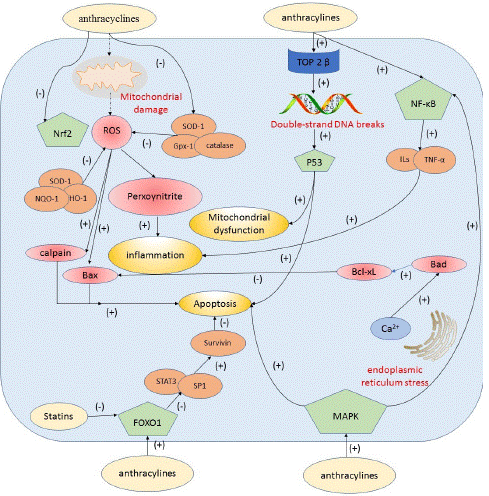
Figure 2: Central schematic figure illustrating the mechanism(s) of anthracycline-based cardiotoxicity and the cardioprotective roles of statins.
Another mechanism of Anthracyclines-induced cardiotoxicity is altered fermodulin iron effector binding [33]. Under physiological conditions, there is only a small amount of bioactive free iron in cardiomyocytes and most of the iron ions exist in the form of bound iron through binding to ferritin. Ferritin prevents iron ions from escaping and avoids damage to cells and tissues. Under certain pathological conditions, reducing agents can reduce ferritin, release active Fe2+, which generate highly reactive and highly toxic hydroxyl radicals by participating in the Haber-Weiss reaction, and cause myocardial cell damage [34].
Apoptosis and Autophagy Theory
Oxidative stress results in the release of cytochrome and activation of caspase 3 in mitochondria leads to apoptosis in the myocardium. The result of apoptosis is increased by Bcl/Bax. And Reactive oxygen species generation increases apoptotic level by activating the Apoptosis Signal-Regulating Kinase 1 (ASK1) signaling [35]. DOX elevates the expression of the Death Receptors (DR) such as tumor necrosis factor receptor 1, fas cell surface death receptor, DR4, and DR5 in cardiomyocyte which elevated expression contributes to activation of caspase cascade shepherd to apoptosis [36]. DOX also mediates apoptosis through the enhancement of HSP25, p53, decrease the GATA-4 expression level and p300 degradation [20,37]. During acute or chronic exposure to anthracyclines, cardiac ATP levels decreased, amp/ATP ratio increased, AMPK phosphorylation and its downstream Acetyl CoA Carboxylase (ACC) levels decreased significantly [38]. Studies have shown that AMPK is inhibited α 1 gene (AMPK)α1-/-) can reduce the phosphorylation of p53 related to anthracyclines and increase the acetylation of p53, so as to promote the accumulation of p53. Therefore, AMPK is essential in anthracyclines induced p53 dependent DNA damage and apoptosis [39]. In addition, anthracyclines can also block the activity of histone deacetylase, increase the acetylation of p53 and increase cell apoptosis.
Autophagy is a mechanism for the degradation of intracellular proteins and organelles mediated by lysosomes, which plays an important physiological part in promoting the metabolism of intracellular substances and energy and maintaining cellular homeostasis. Autophagy preserves cardiac structure and function under baseline conditions and is activated during stress, limiting damage under most conditions. It also reduces injury and preserves cardiac function during ischemia, relieves chronic ischemic remodeling and mediates the cardiac adaptation to pressure overload by restricting mis folded protein accumulation, mitochondrial dysfunction and oxidative stress. Impairment of autophagy is involved in the development of anthracyclines-induced cardiomyopathy [40]. Autophagy has a dual effect in the occurrence and development of cardiotoxicity. Some argue that anthracyclines lead to cardiotoxicity by upregulating cardiac autophagy, while others support that inhibition of cardiac autophagy ultimately leads to cardiotoxicity [41]. To sum up, either autophagy inhibition or over activation impairs cardiac function [42]. On the one hand, anthracyclines may inhibit autophagy by injuring lysosomes, activating mTOR1 and then inhibiting the activities of ULK1 and AMPK. On the other hand, it can inhibit GATA-binding protein 4, leading to the up-regulation of autophagic flux such as Beclin-1, p62 and so on [43], and then result in excessive activation of autophagy and causing myocardial injury [44,45]. A study found that the effect of anthracyclines on autophagy may depend on the dose and duration of treatment. Autophagic fluxes play a key role in various stages of autophagy, and their elevations can reflect enhanced autophagosome formation or blocked autophagosome-lysosome fusion. When autophagy is damaged by anthracycline, autophagy fluxes get corrected mitigates anthracyclines-induced cardiotoxicity [46].
Immune Response and Fibrosis
Toll Like Receptor (TLR) is another cell membrane receptor closely related to anthracyclines-induced cardiotoxicity. It can initiate innate immune response to damaged or dead cell components and participate in anthracyclines mediated cell stress and injury. Cardiomyopathy induced by the DOX has an increased oxidative stress associated with increased TLR2, which induces the NF-κB ultimately leading to apoptosis. And DOX induces the release of a variety of cytokines. It further stops the natural killer cell activity, stimulates the responses of cytotoxic T lymphocytes and decreases the differentiation of macrophages. Collective changes in an immune cell affect the cardiac function during DOX treatment [20]. There is an interstitial fibrosis is observed during DOX cardiomyopathy [47]. DOX inhibits the collagen transcription and translation in tumor cells and elevates expression of TGF-β and phosphor-SMAD3 along with increased collagen deposition, an increment of fibroblast to myofibroblast phenotypic transformation and pro-fibrotic signaling pathway [48]. The study found that Cardiac Anchoring Repeat Protein (CARP) is a structural protein to maintain sarcomere integrity, which expression is regulated by GATA-4, a regulator of cardiomyocyte survival. DOX can induce the consumption of GATA-4 in cardiomyocytes, inhibit CARP transcription, and cause sarcomere disorder, leading to cardiac dysfunction [49]. Treatment of cultured adult rat cardiomyocytes with varying doses of doxorubicin for a short period resulted in a decrease in the elastic properties of titin, and degradation of the protein [50]. Taken together, anthracyclines can disrupt systolic and diastolic cardiac function by effect on cardiac structural integrity. TLR4 can cause cardiac inflammatory response by inhibiting the transcription factor GATA4. Anthracyclines related myocardial injury in TLR4 gene knockout (TLR4-/-) mice is mild [51].
NRG-1/ErbB Pathway and its Application β Adrenergic Receptor
Another mechanism of anthracyclines-induced cardiotoxicity is related to its inhibition of Neuregulin-1 (NRG-1)/ErbB signaling pathway in cardiomyocytes. ErbBs mainly include ErbB1, ErbB2, ErbB3 and ErbB4. They can form dimers or heterodimers on the serosa and can be activated by binding with growth factor ligands. At first, ErbBs was found on the surface of some tumor cells, especially on the surface of breast cancer cells. Zhao showed that ErbB2 and ErbB4 were also expressed on the surface of cardiac myocytes. Recent studies have shown that NRG-1/ErbB is also involved in anthracyclines related cardiotoxicity. Liu's study suggests that single gene knockout heterozygous individuals (NRG-1+/-) can aggravate anthracyclines dependent heart failure and increased mortality. The protective effect of NRG-1 disappeared after inhibiting ErbB2, ErbB4, PI3K, Akt or mTOR [52]. In addition, ErbB2 can up regulate antioxidant enzymes such as glutathione peroxidase 1, reduce the production of oxygen free radicals and protect the heart. ErbB2 knockout mouse cardiomyocytes are more sensitive to anthracyclines induced cell death. Anthracyclines can inhibit the expression of ErbB4 by up regulating mir146a, thereby inhibiting the myocardial protective effect of ErbBs [53]. The above mechanism also explains why the combination of anthracyclines and anti ErbB antibody (trastuzumab) can significantly increase the risk of cardiac dysfunction.
Statins in Anthracyclines-Induced Cardiotoxicity
Anthracyclines related cardioprotective strategies include the following [6]: (1) using a non-anthracyclines regimen if noninferior; (2) administration of cardioprotective agents for the purpose of primary or secondary prevention; (3) continuous infusion or liposomal formulations of anthracyclines to reduce the anthracyclines dose to the heart. The use of dexrazoxane for the prevention of anthracyclines-induced chronic HF restricted by the FDA in 2011, alternative cardio-preventive measures are urgently required. Today cardioprotective preventive measures applied are limitation of the cumulative anthracyclines dose and application of ‘less cardiotoxic’ derivatives or liposomal encapsuled forms. The primary or secondary prevention includes ACE inhibitors, beta-blockers, diuretics, aldosterone blockers and cardiac glycosides [54]. The therapeutic benefit closely depends on the improvement of left ventricular function. A preventive treatment with statins is promising pharmaceutical approaches to alleviate anthracyclines-induced cardiotoxicity. Table 1 shows that statins and their clinical benefits in anthracyclines-induced cardiotoxicity. At the same time, two randomized trials of atorvastatin versus placebo started prior to anthracyclines treatment are ongoing [6], which will provide effective guidance for our clinical treatment. In a word, the direct and indirect actions of statins contribute to an overall reduction of cardiovascular risk.
![]()
Population
Types of Tumors
Types of Expriments
Types of Cardiotoxicity
Follow-up
Erwin Danil Yulian et al.2021 [55]
30 patients receiving simvastatin and 30 patients receiving placeblo
locally advanced breast cancer
RCT
Reductions in heart function were observed in 3.3% cases of simvastatin group and 10% of placebo group.
The treatment cycles were repeated every 3 weeks. three cycles
Sinziana Seicean et al.2012 [56]
67 patients receiving
Statins therapy and 134 patients not receiving statinsBreast Cancer
Retrospective study
4 cases of HF in patients treated with statins compared with 23 cases in the control group
2.55±1.68 years
Husam Abdel‐Qadir et al.2021 [57]
666 patients receiving
Statins therapy and 666 patients not receiving statinsEarly Breast Cancer
Cohort Study
The cumulative incidence of HF hospital presentation was 1.2% (0.5%, 2.6%) in statins‐exposed women, compared with 2.9% (1.7%, 4.6%) in unexposed women. The cause‐specific HR was 0.45 (0.24, 0.85) for statins‐exposed relative to unexposed women (P=0.01).
5 years
Maryam Nabati et al.2018 [58]
38 patients receiving
rosuvastatin therapy and 39 patients receiving placeboBreast Cancer
RCT
An LVEF<45% was seen in 6 patients in the placebo group and 4 patients in the intervention group. Compared with the intervention group, the LVEF was significantly reduced in the placebo group at the end of the study (intergroup P=.012).
2 years
Acar et al.2011 [59]
20 patients receiving
atorvastatin therapy and 20 patients not receiving atorvastatinNon-Hodgkin’s
lymphoma, multiple myeloma
and leukemiaRCT
No difference was observed in the mean EF of the atorvastatin group (61.3±7.9% vs. 62.6±9.3%, p=0.144). the decrease in the control group was significant (62.9±7.0% vs.55±9.5%, p<0.0001)
6 months
Runyawan et al.2015 [60]
14 patients receiving
Statins therapy and 37 patients not receiving statinsBreast cancer, leukemia,
lymphomaCohort study
An increase in LVEF in those receiving a statin
dose of 40-80 mg/d, whereas there was a 3.4%±4% decrease in LVEF after low-dose (10-20 mg/d) statins use and a 9.2%±3% decrease in LVEF in those not receiving statins (P=0.02)6 months
Table 1: Statins and their clinical benefits in anthracyclines-induced cardiotoxicity.
Mechanisms of Statins Alleviating Cardiotoxicity
Statins, 3-Hydroxy-3-Methylglutaryl-Coenzyme A(HMG-CoA) reductase inhibitors, are accepted first line agents to reduce the risk of adverse cardiovascular events, which lower serum cholesterol by inhibiting HMG-CoA reductase to limit cholesterol biosynthesis and enhance low-density lipoprotein clearance from the circulation. Beyond lipid lowering properties in anthracyclines-induced cardiotoxicity, statins are still suggested to have several pleiotropic effects including decreasing oxidative stress and inflammation, improving endothelial function, enhancing stability of atherosclerotic plaques, decreasing platelet activation, inhibiting thrombosis, and inhibition of smooth muscle proliferation [61,62]. This review is proposed to investigate the cardioprotective potential of statins on anthracyclines-induced cardiotoxicity.
Improving Anti-Tumor Response
Feleszko and his colleagues demonstrated that significantly increased sensitivity to the combined treatment with both lovastatin and DOX as compared with either agent acting alone in three tumor models (Co-ion-26 cells, v-Ha-ras-transformed NIH-3T3 sarcoma cells, and Lewis lung carcinoma cells) in vivo. Lovastatin treatment also resulted in a reduction of troponin T release by cardiomyocytes in DOX-treated mice [63], which means that statins foster a win–win situation in anthracyclines-based therapy: sensitizing tumor cells while protecting the heart. In fact, statins sensitize different tumor entities against various chemotherapeutics in rodent models.
Reducing Endothelial Dysfunction
Endothelial dysfunction is the main mechanism of anthracyclines-induced cardiotoxicity. Studies have shown that statins can improve endothelial function through the following mechanisms, thereby exerting many favorable effects on anthracyclines-induced cardiotoxicity [64].
(1) Endothelial NO inhibits several components of the atherogenic process. Such as mediating vascular relaxation [65], inhibiting platelet aggregation and vascular smooth muscle proliferation [66,67] and endothelial–leukocyte interactions [68,69]. In the process of inhibiting cholesterol synthesis, statins inhibit the synthesis of isoprenoid intermediates Farnesyl Pyrophosphate (FPP) and Geranyl Pyrophosphate (GGPP). FPP and GGPP are important components of Rho protein. The active form of Rho can reduce eNOS messenger ribonucleic acid (mRNA) stability and eNOS phosphorylation, resulting in down-regulation of eNOS expression and reduced eNOS activity [70]. Statins improve eNOS mRNA stability by inhibiting prenylation of Rho [70]. Statins can increase endothelial NO production by stimulating and upregulating endothelial NO synthase [71,72] and restore eNOS activity in the presence of hypoxia and oxidized LDL cholesterol [73]. Activation of eNOS is mediated by the serine-threonine protein kinase Akt. Statins promote the rapid activation of the Phosphatidylinositol 3-Kinase/Protein Kinase Akt (PI3K/Akt) pathway, leading to phosphorylation of eNOS to accelerate vascular structure formation [72,74,75]. Statins-mediated upregulation of eNOS is associated with downregulation of markers of platelet reactivity, and potential additional mechanisms include a reduction in the production of thromboxane A2 and modifications in the cholesterol content of platelet membranes [64,76]. Statins also downregulate the expression of caveolin-1 in endothelial cells, a molecule that regulates eNOS subcellular localization and inactivates eNOS [77].
(2) Vascular progenitor cells as smooth muscle progenitor cells and endothelial progenitor cells present in the bone marrow, which are important cell sources involved in the repair of injured blood vessels. The former can differentiate into smooth muscle cells, promote neointima formation and lead to lumen narrowing. The latter can differentiate into endothelial cells, rebuild luminal endothelial cell monolayer and inhibit the remodeling of injured blood vessels. Statins promote the activation of the PI3K/Akt pathway to mobilize endothelial progenitor cells from the bone marrow to accelerate vascular structure formation as well as inhibit the development of neointima. Inhibition of smooth muscle progenitor cell proliferation by statins can reduce excessive neointimal hyperplasia after vascular injury, inhibit restenosis [72,78,79]. Recent studies showed that inhibition of Rho by statins is the key mechanism by which statins inhibit vascular smooth muscle proliferation [64]. Statins also increase the expression of tissue-type plasminogen activator and inhibit the expression of endothelin-1, a potent vasoconstrictor and mitogen [80,81]. Study found that atorvastatin is able to inhibit blood coagulation through several important steps of the coagulation and fibrinolytic system, enhancing thrombolytic capacity and subsequently reducing thrombosis [82].
(3) Anthracyclines also reduce claudin zone occlusive ZO-1 in endothelial cells, thereby increasing microvascular permeability [19]. Statins treatment of human microvascular endothelial cells stimulated activation of ERK5 and translocation to the plasma membrane resulting in co-localisation with the tight junction protein ZO-1 and a concomitant reduction in endothelial cell permeability. Statins pretreatment could overcome the effect of DOX in reducing endothelial tight junction formation and prevent increased permeability [83]. In addition, statins improve endothelial function via blocking recruitment of inflammatory leukocytes and balancing the VEGFa/ANG-PT1 expression, consequently improving left ventricular outcome [84].
Reducing Inflammation and Oxidative Stress
Inflammation and oxidative stress always run through cardiovascular disease. A Study showed rosuvastatin might reduce DOX-induced cardiotoxicity during the early period by blocking ROS formation and reduced oxidative stress resulted in preservation of LV function during the later period [85]. Statins may improve endothelial function through their antioxidant effects, including lipid lowering by itself [86], attenuating angiotensin II–induced free radical production by inhibiting Rac1-mediated nicotinamide adenine dinucleotide oxidase activity and angiotensin-1 receptor expression [87].
The pleiotropic effects of statins are mainly mediated by inhibition of isoprenoids, which serve as lipid attachments for intracellular signaling molecules. HMG-CoA is the rate-limiting enzyme in the mevalonate pathway. Mevalonate is essential for the formation of isoprenoid intermediates that are important for membrane translocation and activation of small Guanosine Triphosphate (GTP) binding proteins, including Rho, Ras, and Rac and may play an important role in mediating the pleiotropic effects of statins [88,89]. Rho and its downstream effector- Rho Kinase (ROCK) regulate the actin cytoskeleton and therefore affect intracellular transport, gene transcription, and messenger RNA expression and stability [90]. Cause increased ROCK activity is associated with endothelial dysfunction, cerebral ischemia, coronary vasospasms and metabolic syndrome, the inhibition of ROCK by statins leads to up-regulation of eNOS, decreased vascular inflammation and reduced atherosclerotic plaque formation [91]. High dose atorvastatin rapidly inhibits the proatherogenic Rho/ROCK pathway, independent of cholesterol reduction, contributes to subsequent reductions in NF-κB signaling partly and enhances the activity of endothelial nitric oxide synthase and increases NO bioavailability stimulating troponin-1 phosphorylation and myocardial relaxation [92,93]. This inhibition may contribute to the clinical benefits of statins, providing a useful therapeutic target in patients with atherosclerosis [94]. Furthermore, statins are known to affect TGF/SMAD/CTGF signaling as well as STAT signaling, both of which play important roles in fibrosis and cancer development [95]. Statins also can inhibit Rho translocation by inhibiting the prenylation of Rho, who is the key factor regulating NADPH oxidase (NOX1) and TOP2 [29]. NOX1 is an important enzyme generating ROS in vascular endothelial cells and macrophages, while Rac1 is part of the NOX1 and can promote the inflammatory process. Therefore, atorvastatin attenuates downstream NOX1 and NOX2 activity by inhibiting Rac1-mediated NADPH activity, and subsequently suppresses ROS generation and release [91,96]. It is speculated that statins may resist cardiomyocyte damage by inhibiting Rac1-driven prooxidative mechanisms [97]. Atorvastatin treatment combined with DOX treatment led to a significant reduction in the oxidative stress, presenting a significant decrease in the lipid peroxidation levels and increase in the glutathione levels in the heart and testes after short- and long-term treatment [98]. It was shown that atorvastatin also can activate Erk5, an external signaling regulatory enzyme of the MAPK signaling pathway, to downregulate the expression of the protein Rac1 and reduce the levels of VCAM-1 and ICAM-1 to reduce inflammation [99].
Fluvastatin pretreatment has been reported to attenuate DOX induced cardiotoxicity via reducing cardiac expression of nitrotyrosine, enhancing expression of the mitochondrial located antioxidative SOD2, attenuating mitochondrial apoptotic pathways and reducing cardiac inflammatory response [100]. Simvastatin ameliorated DOX induced nephropathy in rat via its anti-inflammatory action through a reduction of NF-κB activation, and IL-1β and TGF-β expression [101]. Since NF-κB functions as a pro-inflammatory transcription factor and regulates in a Rho-dependent manner, it is feasible for statins to interfere with anthracyclines-induced cardiac inflammation by inhibiting NF-κB signaling as well as interfering with NOX [102]. Meanwhile, statins can increase the expression of Sirt1, further inhibit the activity of NF-κB, terminate the release of downstream inflammatory mediators, and jointly protect the myocardial toxic injury of DOX [103]. Note worthy, certain statins show anti-oxidative capacity such as atorvastatin, simvastatin and Fluvastatin, who can activate Keap1/Nrf2 signaling [104,105]. Nrf2 is responsible for the expression of genes coding for anti-oxidative factors, including NAD(P)H dehydrogenase quinone oxidoreductase 1 (NQO1), Myocardial Heme Oxygenase-1(HO-1), Glutathione Reductase (GRX1) and Cu/Zn-SOD [106]. Nrf2 has also been shown to protect the heart by removing toxic ubiquitinated protein aggregates produced by ROS by autophagy.
Study indicated Cyclooxygenase (COX)-2 is an inducible pro-inflammatory enzyme with important effects in increasing inflammation, angiogenesis and atherosclerotic plaques, whose underlying molecular mechanisms are complex and not fully explained. But the occurrence of a functional relationship between COX-2 activity and the release of Metalloproteinases (MMPs) has been demonstrated in several studies to play a key importance in it [107]. Insight into the regulation of COX-2 and MMP-9 expression suggests the involvement of a Rho-dependent pathway. Statins interfere with Rho activation and thereby reduce COX-2 and MMP-9 expression and activity. In a word, statins hold the potential to ameliorate Anthracyclines-induced pro-oxidative, inflammatory and fibrotic cardiac responses through inhibition of Rho-GTPase signaling.
Inhibiting DNA Damage and Apoptosis
A large body of evidence supports the notion that cardiac myocyte death by apoptosis and necrosis is a primary mechanism of Anthracyclines-induced cardiotoxicity. A study demonstrated the cardioprotective potential of rosuvastatin against DOX-induced myocardial apoptosis [108]. Survivin, one member of inhibitor of apoptosis protein family, encoded by BIRC5, plays an important role in regulating apoptosis and cell division, which exert an anti-apoptotic effect against Anthracyclines-induced cardiotoxicity [109,110]. Survivin has a potentially cytoprotective effect against DOX-induced cardiac myocyte apoptosis through mechanisms that involve a decrease in the phosphorylation of p38 MAP kinase, mitochondrial Smac release, and increased expression of Bcl-2 and CREB [110]. The FOXO transcription factor family (FOXO1, FOXO3, FOXO4, FOXO6) belongs to the winged helix or forkhead box class of transcription factors [111]. FOXO1 by protein phosphorylation and inhibition of nuclear translocation allowing for the inhabition of anthracyclines-induced cardiotoxicity [112]. The transcriptional regulation of survivin is mediated via this FOXO1/STAT3/Sp1 transcriptional network. Clinically, statins uses are associated with a lower risk for HF in breast cancer patients with anthracyclines chemotherapy. Sp1 was a critical transcription factor in transcriptional regulation of survivin in cardiomyocytes, especially for DOX-induced toxicity [113]. In vitro studies showed that DOX treatment resulted in the activation of FOXO1 protein which competitively inhibits STAT3 from binding to Sp1, and then decrease expression of the apoptotic inhibitor protein. Pre-treatment with statins significantly inhibit FOXO1 binding to STAT3 and restores STAT3 binding to Sp1 and stabilized transcription complex of STAT3/Sp1. The study suggested a new pathophysiologic mechanism that survivin mediated protective effect of atorvastatin against DOX-induced cardiotoxicity via FOXO1/STAT3/Sp1 transcriptional network [114].
In mice, lovastatin mitigated acute DOX induced heart damage as indicated by reduced mRNA levels of the pro-fibrotic cytokine Connective Tissue Growth Factor (CTGF) and pro-inflammatory cytokines. Lovastatin also protected from DOX-provoked subacute cardiac damage as shown by lowered mRNA levels of CTGF and atrial natriuretic peptide. Increase in the serum concentration of troponin I and cardiac fibrosis following DOX treatment were also reduced by lovastatin [115]. Lovastatin protected the human endothelial cells against DOX-induced toxicity by impairing DNA strand break formation [116]. So does Atorvastatin [2]. The predominant role of Rac1 in the promotion of DOX-induced DSB formation was confirmed in experiments using the Rac1-specific inhibitor [117]. In some studies, the protective effects of statins, such as lowering DSBs levels, attenuating DNA damage response, and reducing apoptotic cell death, have been simulated by using small molecule inhibitors or specific inhibition of Rac1 by clostridium toxins [115,118]. The connection between Rac1 and type II topoisomerases is uncertain but underpinned by findings of Sandrock et al, who showed nuclear Rac1 to co-precipitate with type II topoisomerases [119]. Moreover, mice with liver-specific rac1 knockout are protected from acute anthracyclines-triggered DSB formation in hepatocytes [120]. Chronic DOX-induced cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity [121].
Conclusion
Statins combined with anthracyclines are feasible strategies for reducing cardiotoxicity in antitumor therapy. Statins not only protect heart function by increasing LEVF and reducing the incidence of HF, but also could it increase the sensitivity of some tumors to chemotherapy drugs and improve the anti-tumor efficiency of anthracyclines, while protecting normal tissue, further widening the treatment window. In a word, statins have the potential to alleviate acute, sub-acute and chronic cardiotoxicity following anthracyclines treatment. Several studies have shown that the beneficial effects of statins come from inhibition of RhoA or Rac1 signaling, making Rho GTP enzymes a preferred target for future chemoprevention strategies. To date, there is insufficient evidence to recommend the routine use of statins for the primary prevention of anthracyclines cardiotoxicity, the efficacy of statins in reducing the anthracyclines-induced cardiotoxicity has been validated in small-scale clinical trials. However, the cardioprotective effects of statins in anthracyclines still require further validation in large-scale randomized prospective studies.
References
- Saleh Y, Abdelkarim O, Herzallah K, Abela GS. Anthracycline-induced cardiotoxicity: mechanisms of action, incidence, risk factors, prevention, and treatment. Heart failure reviews. 2021; 26: 1159-73.
- Ramanjaneyulu SV, Trivedi PP, Kushwaha S, Vikram A, Jena GB. Protective role of atorvastatin against doxorubicin-induced cardiotoxicity and testicular toxicity in mice. Journal of physiology and biochemistry. 2013; 69: 513-25.
- Narayan HK, French B, Khan AM, Plappert T, Hyman D, et al. Noninvasive Measures of Ventricular-Arterial Coupling and Circumferential Strain Predict Cancer Therapeutics-Related Cardiac Dysfunction. JACC Cardiovascular imaging. 2016; 9: 1131-41.
- van Nimwegen FA, Schaapveld M, Janus CP, Krol AD, Petersen EJ, et al. Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA internal medicine. 2015; 175: 1007-17.
- Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). European heart journal. 2016; 37: 2768-801.
- Upshaw JN. Cardioprotective Strategies to Prevent Cancer Treatment-Related Cardiovascular Toxicity: a Review. Current oncology reports. 2020; 22: 72.
- Tan C, Tasaka H, Yu KP, Murphy ML, Karnofsky DA. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer. 1967; 20: 333-53.
- Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, et al. Early Detection of Anthracycline Cardiotoxicity and Improvement With Heart Failure Therapy. Circulation. 2015.
- Mulrooney DA, Yeazel MW, Kawashima T, Mertens AC, Mitby P, et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ (Clinical research ed). 2009; 339: b4606.
- Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003; 97: 2869-79.
- Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015; 131: 1981-8.
- Bernstein D. Anthracycline Cardiotoxicity: Worrisome Enough to Have You Quaking? Circulation research. 2018; 122: 188-90.
- Herrmann J, Lerman A, Sandhu NP, Villarraga HR, Mulvagh SL, et al. Evaluation and management of patients with heart disease and cancer: cardio-oncology. Mayo Clinic proceedings. 2014; 89: 1287-306.
- Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. Journal of the American College of Cardiology. 2010; 55: 213-20.
- Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nature medicine. 2012; 18: 1639-42.
- Beretta GL, Zunino F. Molecular mechanisms of anthracycline activity. Topics in current chemistry. 2008; 283: 1-19.
- Zhang H, Zhang YW, Yasukawa T, Dalla Rosa I, Khiati S, et al. Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases IIα and IIβ in vertebrate mitochondria. Nucleic acids research. 2014; 42: 7259-67.
- Thayer WS. Adriamycin stimulated superoxide formation in submitochondrial particles. Chemico-biological interactions. 1977; 19: 265-78.
- Hsu PY, Mammadova A, Benkirane-Jessel N, Désaubry L, Nebigil CG. Updates on Anticancer Therapy-Mediated Vascular Toxicity and New Horizons in Therapeutic Strategies. Frontiers in cardiovascular medicine. 2021; 8: 694711.
- Renu K, V GA, P BT, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy - An update. European journal of pharmacology. 2018; 818: 241-53.
- Bai P, Mabley JG, Liaudet L, Virág L, Szabó C, et al. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncology reports. 2004; 11: 505-8.
- Simůnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, et al. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacological reports: PR. 2009; 61: 154-71.
- Nitiss JL. DNA topoisomerases in cancer chemotherapy: using enzymes to generate selective DNA damage. Current opinion in investigational drugs (London, England: 2000). 2002; 3: 1512-6.
- Marinello J, Delcuratolo M, Capranico G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. International journal of molecular sciences. 2018; 19: 3480.
- Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nature reviews Cancer. 2016; 16: 20-33.
- Mordente A, Meucci E, Silvestrini A, Martorana GE, Giardina B. Anthracyclines and mitochondria. Advances in experimental medicine and biology. 2012; 942: 385-419.
- Jirkovsky E, Popelová O, Kriváková-Stanková P, Vávrová A, Hroch M, et al. Chronic anthracycline cardiotoxicity: molecular and functional analysis with focus on nuclear factor erythroid 2-related factor 2 and mitochondrial biogenesis pathways. The Journal of pharmacology and experimental therapeutics. 2012; 343: 468-78.
- Henninger C, Huelsenbeck S, Wenzel P, Brand M, Huelsenbeck J, et al. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacological research. 2015; 91: 47-56.
- Henninger C, Fritz G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell death & disease. 2017; 8: e2564.
- M RV, Filippelli A, Rinaldi B, Rossi S, Palazzo E, et al. Effects of docosahexaenoic acid on [Ca(2+)](i) increase induced by doxorubicin in ventricular rat cardiomyocytes. Life sciences. 2002; 71: 1905-16.
- Santos DL, Moreno AJ, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicology and applied pharmacology. 2002; 185: 218-27.
- Yamaguchi O, Murakawa T, Nishida K, Otsu K. Receptor-mediated mitophagy. Journal of molecular and cellular cardiology. 2016; 95: 50-6.
- Cusack BJ, Gambliel H, Musser B, Hadjokas N, Shadle SE, et al. Prevention of chronic anthracycline cardiotoxicity in the adult Fischer 344 rat by dexrazoxane and effects on iron metabolism. Cancer chemotherapy and pharmacology. 2006; 58: 517-26.
- Fang X, Wang H, Han D, Xie E, Yang X, et al. Ferroptosis as a target for protection against cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 2019; 116: 2672-80.
- Gilleron M, Marechal X, Montaigne D, Franczak J, Neviere R, et al. NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochemical and biophysical research communications. 2009; 388: 727-31.
- Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Scientific reports. 2017; 7: 44735.
- Tan TC, Neilan TG, Francis S, Plana JC, Scherrer-Crosbie M. Anthracycline-Induced Cardiomyopathy in Adults. Comprehensive Physiology. 2015; 5: 1517-40.
- Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chaté V, Schnebelen C, et al. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovascular research. 2012; 95: 290-9.
- Wang S, Song P, Zou MH. Inhibition of AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. The Journal of biological chemistry. 2012; 287: 8001-12.
- Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The Role of Autophagy in the Heart. Annual review of physiology. 2018; 80: 1-26.
- Li M, Russo M, Pirozzi F, Tocchetti CG, Ghigo A. Autophagy and cancer therapy cardiotoxicity: From molecular mechanisms to therapeutic opportunities. Biochimica et biophysica acta Molecular cell research. 2020; 1867: 118493.
- Yamaguchi O. Autophagy in the Heart. Circulation journal: official journal of the Japanese Circulation Society. 2019; 83: 697-704.
- Zhang QL, Yang JJ, Zhang HS. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019; 109: 71-83.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018; 14: 207-15.
- Li M, Sala V, De Santis MC, Cimino J, Cappello P, et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation. 2018; 138: 696-711.
- Moriyama S, Fukata M, Kusaba H, Maruyama T, Akashi K. Acute and Chronic Effects of Cancer Drugs on the Cardiovascular System. Heart failure clinics. 2020; 16: 231-41.
- Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, etal. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Medicinal research reviews. 2014; 34: 106-35.
- Oliveira MS, Carvalho JL, Campos AC, Gomes DA, de Goes AM, et al. Doxorubicin has in vivo toxicological effects on ex vivo cultured mesenchymal stem cells. Toxicology letters. 2014; 224: 380-6.
- Chen B, Zhong L, Roush SF, Pentassuglia L, Peng X, et al. Disruption of a GATA4/Ankrd1 signaling axis in cardiomyocytes leads to sarcomere disarray: implications for anthracycline cardiomyopathy. PloS one. 2012; 7: e35743.
- Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, et al. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. The Journal of biological chemistry. 2004; 279: 8290-9.
- Riad A, Bien S, Gratz M, Escher F, Westermann D, et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. European journal of heart failure. 2008; 10: 233-43.
- Fukazawa R, Miller TA, Kuramochi Y, Frantz S, Kim YD, et al. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. Journal of molecular and cellular cardiology. 2003; 35: 1473-9.
- Horie T, Ono K, Nishi H, Nagao K, Kinoshita M, et al. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovascular research. 2010; 87: 656-64.
- Volkova M, Russell R, 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Current cardiology reviews. 2011; 7: 214-20.
- Yulian ED, Siregar NC, Bajuadji. Combination of Simvastatin and FAC Improves Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer. Cancer research and treatment. 2021; 53: 1072-83.
- Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. Journal of the American College of Cardiology. 2012; 60: 2384-90.
- Abdel-Qadir H, Bobrowski D, Zhou L, Austin PC, Calvillo-Argüelles O, et al. Statin Exposure and Risk of Heart Failure After Anthracycline- or Trastuzumab-Based Chemotherapy for Early Breast Cancer: A Propensity Score‒Matched Cohort Study. Journal of the American Heart Association. 2021; 10: e018393.
- Nabati M, Janbabai G, Esmailian J, Yazdani J. Effect of Rosuvastatin in Preventing Chemotherapy-Induced Cardiotoxicity in Women With Breast Cancer: A Randomized, Single-Blind, Placebo-Controlled Trial. Journal of cardiovascular pharmacology and therapeutics. 2019; 24: 233-41.
- Acar Z, Kale A, Turgut M, Demircan S, Durna K, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. Journal of the American College of Cardiology. 2011; 58: 988-9.
- Chotenimitkhun R, D’Agostino R Jr, Lawrence JA, Hamilton CA, Jordan JH, et al. Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. The Canadian journal of cardiology. 2015; 31: 302-7.
- Kunutsor SK, Laukkanen JA. Heart Failure Risk Reduction: Hydrophilic or Lipophilic Statins? Cardiology. 2020; 145: 384-6.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. The American journal of cardiology. 2005; 96: 24F-33F.
- Feleszko W, Mlynarczuk I, Balkowiec-Iskra EZ, Czajka A, Switaj T, et al. Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000; 6: 2044-52.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. The American journal of cardiology. 2005; 96: 24f-33f.
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 1987; 84: 9265-9.
- Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. British journal of pharmacology. 1992; 107: 745-9.
- Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. The Journal of clinical investigation. 1989; 83: 1774-7.
- Gauthier TW, Scalia R, Murohara T, Guo JP, Lefer AM. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 1995; 15: 1652-9.
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proceedings of the National Academy of Sciences of the United States of America. 1991; 88: 4651-5.
- Zhou Q, Liao JK. Pleiotropic effects of statins. - Basic research and clinical perspectives. Circulation journal: official journal of the Japanese Circulation Society. 2010; 74: 818-26.
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109: Iii27-32.
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature medicine. 2000; 6: 1004-10.
- Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. The Journal of biological chemistry. 1997; 272: 31725-9.
- Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. The Journal of clinical investigation. 2001; 108: 391-7.
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nature medicine. 2003; 9: 1370-6.
- Vaughan CJ, Gotto AM Jr, Basson CT. The evolving role of statins in the management of atherosclerosis. Journal of the American College of Cardiology. 2000; 35: 1-10.
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001; 103: 113-8.
- Lavi R, Zhu XY, Chade AR, Lin J, Lerman A, et al. Simvastatin decreases endothelial progenitor cell apoptosis in the kidney of hypertensive hypercholesterolemic pigs. Arteriosclerosis, thrombosis, and vascular biology. 2010; 30: 976-83.
- Schaefer CA, Kuhlmann CR, Gast C, Weiterer S, Li F, et al. Statins prevent oxidized low-density lipoprotein- and lysophosphatidylcholine-induced proliferation of human endothelial cells. Vascular pharmacology. 2004; 41: 67-73.
- Essig M, Nguyen G, Prié D, Escoubet B, Sraer JD, et al. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circulation research. 1998; 83: 683-90.
- Hernández-Perera O, Pérez-Sala D, Navarro-Antolín J, Sánchez-Pascuala R, Hernández G, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. The Journal of clinical investigation. 1998; 101: 2711-9.
- Fenton JW 2nd, Brezniak DV, Ofosu FA, Shen GX, Jacobson JR, et al. Statins and thrombin. Current drug targets Cardiovascular & haematological disorders. 2005; 5: 115-20.
- Wilkinson EL, Sidaway JE, Cross MJ. Statin regulated ERK5 stimulates tight junction formation and reduces permeability in human cardiac endothelial cells. Journal of cellular physiology. 2018; 233: 186-200.
- Leenders GJ, Smeets MB, van den Boomen M, Berben M, Nabben M, et al. Statins Promote Cardiac Infarct Healing by Modulating Endothelial Barrier Function Revealed by Contrast-Enhanced Magnetic Resonance Imaging. Arteriosclerosis, thrombosis, and vascular biology. 2018; 38: 186-94.
- Kim YH, Park SM, Kim M, Kim SH, Lim SY, et al. Cardioprotective effects of rosuvastatin and carvedilol on delayed cardiotoxicity of doxorubicin in rats. Toxicology mechanisms and methods. 2012; 22: 488-98.
- Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-Kinase in the Cardiovascular System. Circulation research. 2016; 118: 352-66.
- Wassmann S, Laufs U, Bäumer AT, Müller K, Ahlbory K, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension (Dallas, Tex: 1979). 2001; 37: 1450-7.
- Muranaka T. Regulation of the Mevalonate Pathway. Chemical Regulation of Plants. 1997; 32: 199-210.
- Takemoto M, Liao JK. Pleiotropic Effects of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors. Arteriosclerosis Thrombosis & Vascular Biology. 2001; 21: 1712-9.
- Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Experimental Cell Research. 2000; 261: 44-51.
- Zhou Q, Liao JK. Rho kinase: an important mediator of atherosclerosis and vascular disease. Current pharmaceutical design. 2009; 15: 3108-15.
- Heiston EM, Hundley WG. Statins for Cardiac and Vascular Protection During and After Cancer Therapy. Current oncology reports. 2022; 24: 555-561.
- Wei CY, Huang KC, Chou YH, Hsieh PF, Lin KH, et al. The role of Rho-associated kinase in differential regulation by statins of interleukin-1beta- and lipopolysaccharide-mediated nuclear factor kappaB activation and inducible nitric-oxide synthase geneexpression in vascular smooth muscle cells. Molecular pharmacology. 2006; 69: 960-7.
- Nohria A, Prsic A, Liu PY, Okamoto R, Creager MA, Selwyn A, et al. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis. 2009; 205: 517-21.
- Rizvi F, Siddiqui R, DeFranco A, Homar P, Emelyanova L, et al. Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. International journal of cardiology. 2018; 270: 228-36.
- Zhang XZ, Huang X, Qiao JH, Zhang JJ, Zhang MX. Inhibition of hypoxia-induced retinal neovascularization in mice with short hairpin RNA targeting Rac1, possibly via blockading redox signaling. Experimental eye research. 2011; 92: 473-81.
- Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103: 7432-7.
- Prahalathan C, Selvakumar E, Varalakshmi P. Remedial effect of DL-alpha-lipoic acid against adriamycin induced testicular lipid peroxidation. Molecular and cellular biochemistry. 2004; 267: 209-14.
- McGown CC, Brookes ZL, Hellewell PG, Ross JJ, Brown NJ. Atorvastatin reduces endotoxin-induced microvascular inflammation via NOSII. Naunyn-Schmiedeberg’s archives of pharmacology. 2015; 388: 557-64.
- Riad A, Bien S, Westermann D, Becher PM, Loya K, et al. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer research. 2009; 69: 695-9.
- Zhang W, Li Q, Wang L, Yang X. Simvastatin ameliorates glomerulosclerosis in Adriamycin-induced-nephropathy rats. Pediatric nephrology (Berlin, Germany). 2008; 23: 2185-94.
- Guo F, Xing Y, Zhou Z, Dou Y, Tang J, et al. Guanine-nucleotide exchange factor H1 mediates lipopolysaccharide-induced interleukin 6 and tumor necrosis factor α expression in endothelial cells via activation of nuclear factor κB. Shock (Augusta, Ga). 2012; 37: 531-8.
- Lei J, Gu X, Ye Z, Shi J, Zheng X. Antiaging effects of simvastatin on vascular endothelial cells. Clinical and applied thrombosis/hemostasis: official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis. 2014; 20: 212-8.
- Franzoni F, Quiñones-Galvan A, Regoli F, Ferrannini E, Galetta F. A comparative study of the in vitro antioxidant activity of statins. International journal of cardiology. 2003; 90: 317-21.
- Habeos IG, Ziros PG, Chartoumpekis D, Psyrogiannis A, Kyriazopoulou V, et al. Simvastatin activates Keap1/Nrf2 signaling in rat liver. Journal of molecular medicine (Berlin, Germany). 2008; 86: 1279-85.
- van der Zanden SY, Qiao X, Neefjes J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. The FEBS journal. 2021; 288: 6095-111.
- Marika M, Antonella Z, Egeria S, Annunziata CM, Carlo S, et al. Statins inhibit cyclooxygenase-2 and matrix metalloproteinase-9 in human endothelial cells: anti-angiogenic actions possibly contributing to plaque stability. Cardiovascular research. 2010; 86: 311-20.
- Sharma H, Pathan RA, Kumar V, Javed S, Bhandari U. Anti-apoptotic potential of rosuvastatin pretreatment in murine model of cardiomyopathy. International journal of cardiology. 2011; 150: 193-200.
- Athanasoula K, Gogas H, Polonifi K, Vaiopoulos AG, Polyzos A, et al. Survivin beyond physiology: orchestration of multistep carcinogenesis and therapeutic potentials. Cancer letters. 2014; 347: 175-82.
- Lee BS, Kim SH, Jin T, Choi EY, Oh J, et al. Protective effect of survivin in Doxorubicin-induced cell death in h9c2 cardiac myocytes. Korean circulation journal. 2013; 43: 400-7.
- Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. The Journal of biological chemistry. 2011; 286: 7468-78.
- Henninger C, Fritz G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell death & disease. 2017; 8: e2564.
- Lee BS, Oh J, Kang SK, Park S, Lee SH, et al. Insulin Protects Cardiac Myocytes from Doxorubicin Toxicity by Sp1-Mediated Transactivation of Survivin. PloS one. 2015; 10: e0135438.
- Oh J, Lee BS, Lim G, Lim H, Lee CJ, et al. Atorvastatin protects cardiomyocyte from doxorubicin toxicity by modulating survivin expression through FOXO1 inhibition. Journal of molecular and cellular cardiology. 2020; 138: 244-55.
- Huelsenbeck J, Henninger C, Schad A, Lackner KJ, Kaina B, et al. Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell death & disease. 2011; 2: e190.
- Damrot J, Nübel T, Epe B, Roos WP, Kaina B, et al. Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. British journal of pharmacology. 2006; 149: 988-97.
- Wartlick F, Bopp A, Henninger C, Fritz G. DNA damage response (DDR) induced by topoisomerase II poisons requires nuclear function of the small GTPase Rac. Biochimica et biophysica acta. 2013; 1833: 3093-103.
- Huelsenbeck SC, Schorr A, Roos WP, Huelsenbeck J, Henninger C, et al. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. The Journal of biological chemistry. 2012; 287: 38590-9.
- Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. The Journal of pharmacology and experimental therapeutics. 2011; 336: 496-505.
- Bopp A, Wartlick F, Henninger C, Kaina B, Fritz G. Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell death & disease. 2013; 4: e558.
- Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. Journal of molecular and cellular cardiology. 2009; 47: 698-705.