
Abstract
Porphyria Cutanea Tarda (PCT) is a condition characterized by accumulation of the carboxyl group substituent’s uroporphyrin I and heptacarboxyl porphyrin III resulting in increased iron storage and photosensitivity dermatitis. Here we present a case of a 51-year-oldman who presented with bilateral dorsal hand lesions and iron overload toxicity. Further screening revealed PCT resulting from a mutation in Uroporphyrinogen Decarboxylase (UROD) as well as from genetichemochromatosis (HFE) caused by C282Y homozygosity. The patient was treated successfully with phlebotomy.
Keywords: Porphyria Cutanea Tarda (PCT), Hemochromatosis (HFE), C282Y mutation
Abbreviations
PCT: Porphyria Cutanea Tarda; HFE: Hemochromatosis; UROD: Uroporphyrinogen decarboxylase
Introduction
Porphyria is an encompassing term for diseases in which Uroporphyrinogen Decarboxylase (UROD) deficiency results in overproduction of 4-8 carboxyl group substituents. Porphyria Cutanea Tarda (PCT) is characterized by the accumulation of uroporphyrin I, heptacarboxyl porphyrin III and iron overload toxicity. Deposition and photoexcitement of these porphyrins in the skin cause oxidative damage to the surrounding tissues resulting in classical PCT photosensitivity dermatitis. We present a case of a man who presented with sporadic season dependent bilateral cutaneous lesions on his dorsal hands and nailbeds, who was found to have new onset porphyria cutanea tarda as well as C282Y mutation homozygosity resulting in genetichemochromatosis (HFE) and underlying Hepatitis C (HCV) with undetectable viral load.
Case Report
A 51-year-old male with a long-standing history of hepatitis C secondary to past intravenous drug use with undetectable viral load, and diabetes Type II presented to his primary care physician with worsening cutaneous lesions that had been appearing on his hands. During the past 7summershe would experience painful dorsal upper extremities resulting in disfiguration of his hands and nails (Figure 1). The patient’s daily medications were Lisinopril 20mg and Linagliptin 5mg once daily, Metformin 1000mg twice daily and insulin Detemir subcutaneously. He denied any supplemental iron usage or previous erythrocyte transfusions and denied any similar family history. At the time of his initial visit he had been referred to a dermatologist who performed a shave biopsy of the right index finger.
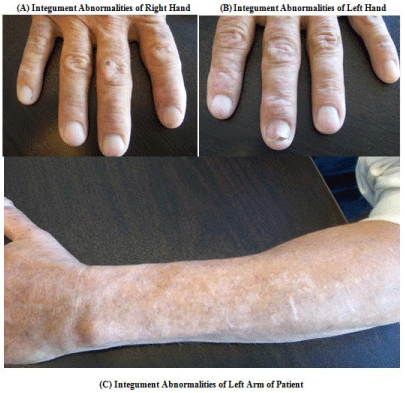
Figure 1: (A) Right hand of patient demonstrating tense bulla with milia and
hyperpigmented macules distributed on the right index finger. (B) Left hand
of patient demonstrating tense bulla with milia and hyperpigmented macules.
(C) Left arm of patient demonstrating hyperpigmented macules.
Physical examination was notable for tense bullae with milia and hyperpigmented macules distributed on the right index finger and left hand. Cardiopulmonary examination was normal and the jugular venous waveform was not elevated. There were no stigmata of chronic liver disease or endocarditis. No peritoneal signs were present. There was no lower extremity edema. The neurological exam was normal. The exam was otherwise normal.
A shave biopsy specimen was taken off the right index finger and sent for manual immunofluorescence staining for IgG, IgM, IgA and Complement 3 which subsequently returned negative. Histopathology revealed subepidermal vesiculobullous disease and colloid bodies.
Subsequent hematologic analysis (Table 1) revealed an elevated ferritin of 1111ng/ml (Normal Range [NR] 30-400ng/ml) and elevated liver enzymes of both AST 80U/L (NR 10–35U/L) and ALT142U/L (NR10–35U/L). Comprehensive metabolic panel and complete blood count were otherwise normal.
![]()
Reference Range
Day –35 Initial Discovery
Day 1
Day 15*
Day 75
Day 89
Ferritin, ng/ml
30-400
1111
986
445
193
Aspartate aminotransferase, U/L
10–35
80
41
Alanine aminotransferase, U/L
10�35
142
74
Hematocrit
38.8-50
46.6 %
46.9 %
Mean Corpuscular Hemoglobin pg/cell
27-33
35.0 pg
33.4 pg
* biweekly phlebotomy of 450ml started
Table 1: Ferritin Panel, Liver enzymes and blood count.
Urinary porphyrin testing (Table 2) revealed significantly elevated Coproporphyrin I 110ug/L (NR 0-15ug/L), Uroporphyrin 1765ug/L (NR 0-20 ug/L), Heptacarboxylporphyrin 1694ug/L (NR 0-2ug/L) Pentacarboxylporphyrin 100ug/L (NR 0-2 ug/L) and Protoporphyrin 53ug/dL (NR 0-1ug/dL). Serum porphyrins (Table 2) were normal with the exception of Heptaporphyrin 16ug/dL, Uroporphyrin 28.4ug/dL and Protoporphyrin 17.4ug/Dl (NR for serum porphyrins are 0-1ug/dL).
![]()
Test
Reference Range
Day 1
Urinary Porphyrin Levels
Coproporphyrin I ug/L
0-15
110
U ALA (Delta) 0.0-5.4 mg/L
0.0-5.4
2.2 mg/L
Uroporphyrin 0-20 ug/L
0-20
1765 ug/L
Heptacarboxylporphyrin 0-2 ug/L
0-2
1694 ug/L
Pentacarboxylporphyrin0-2 ug/L
0-2
100 ug/L
Coproporphyrin III 0-49 ug/L
0-49
7 ug/L
Protoporphyrin (FEP) 0-1 ug/dL
0-1
53 ug/dL
Porphobilinogen 0-2 mg/L
0-2
0.7 mg/L
Protoporphyrin (Zinc)0-100 ug/dL
0-100
58 ug/dL
Hexacarboxylporphyrin 0-1 ug/L
0-1
<1 ug/L
Serum Porphyrin Levels
Porphyrins(Total) ug/dL
50.0 ug/dL
Heptaporphyrin ug/dL
0-1
16.0 ug/dL
Pentaporphyrin ug/dL
0-1
< 1.0 ug/dL
Coproporphyrin ug/dL
0-1
< 1.0 ug/dL
Uroporphyrin ug/dL
0-1
28.4 ug/dL
Hexaporphyrin ug/dL
0-1
< 1.0 ug/dL
Protoporphyrin ug/dL
0-1
17.4 ug/dL
Table 2: Porphyrin Results.
Genetic testing revealed a UROD Type II autosomal dominant mutation and C282Y homozygosity.H63D and S65C gene mutations were negative.AFP levels were negligible.
Management consisted of biweekly phlebotomy of 450ml and minimization of sunlight exposure. Subsequently the patient’s ferritin level dropped to 986ng/ml after the first therapeutic intervention, and after 2.7L of phlebotomized blood was removed the patient’s ferritin further dropped to 193ng/ml (Table 2). Ferritin goal was <25ng/ml.
Discussion
Our patient’s worsening summertime bilateral hand bullae were the presenting symptom for his PCT and underlying HFE. His transferrin receptor dysfunction secondary to HFE led to his high ferritin levels, and increasing liver enzymes pointed toward ferritin toxicity. Left unchecked ferritin toxicity leads to cirrhosis and increase the risk of hepatocellular carcinoma [1].
It comes as no surprise then that diseases effecting iron transport and hepatic function can exacerbate the symptomology of excessive serum porphyrins seen in PCT. Although the exact mechanism by which PCT and HFE exacerbate each other is unknown, there has been a documented increase of C282YHFE incidence in Spanish, Italian and Australian patients with PCT [2-4]. In the United States, HFE can occur in approximately 1 out of 200-500 individuals, most of whom are of Northern European population descent [5]. The prevalence of single allele C282Y mutation in those with HFE in the United States is 5.4%, and homozygosity is even more uncommon at 0.26%. It should be noted however that the majority of genetic HFE originates from the H63D mutation which occurs in 13.5% of HFE patients in the United States [6].
Hemochromatosis is not the only exacerbating factor for patients with PCT. Additionally, ethanol, estrogen, hepatitis as well as human immunodeficiency virus can contribute to decreased hepatic expression of hepcidin. Thisin turn leads to deregulation of iron absorption and metabolism causing eventual iron overload and toxicity [7].
Currently there is no documented PCT incidence in the United States, however other developed countries such as Denmark have found incidence of PCT to be approximately 1 per 200,000 [8]. Although PCT is typically associated with a UROD mutation, familial PCT without detectable UROD mutations exist [9]. PCT also occurs through exposure to polyhalogenated aromatic hydrocarbon hepatotoxins used in agriculture [10]. Thus PCT can be seen in all demographic and age groups.
Diagnosis of PCT is made with dermatologic biopsy of suspicious lesion followed by laboratory testing of serum and urine porphyrins. Pathologic findings are linear eosinophilic globules composed of basement membrane material and degenerating keratinocytes in the blister roof [11].
Phlebotomy is the standard treatment for hemochromatosis, polycythemia vera as well as PCT [12] Over the course of several biweekly phlebotomy sessions of 450ml per session, our patient’s ferritin level decreased significantly as did his abnormally high liver enzymes. If phlebotomy is not an option, a 2012 pilot study using an oral iron chelating agent desferasirox showed 7/10 patients with PCT had resolution of blistering [13] However alternate studies have shown that for patients with anemia of chronic disease on dialysis in end stage renal disease erythropoietin can be utilized to stimulate red blood cell production and decrease iron stores [14].
Unfortunately both PCT and hemochromatosis are associated with an increased risk for hepatocellular carcinoma [15]. A study of patients with PCT in north America reported 56% of patients with PCT had associated hepatitis C. Furthermore, 42% of these patients had at least one C282Y mutation for HFE [16]. These two studies conclude that all patients with diagnosed PCT should be tested for HCV infection, HFE gene mutation and screened for Alpha Fetal Protein (AFP) level for hepatocellular carcinoma. Fortunately, our patient’s AFP levels were negligible, as was his HCV viral load. In patients with significant HCV viral load it has been suggested that initial phlebotomy may assist in mitigating hepatic siderosis and increasing efficacy of hepatitis C therapies [17].
In conclusion PCT remains an uncommon disease in the United States, often delaying clinical diagnosis and therapeutic intervention. Clinicians should be suspicious of new or chronic rashes worsening with sun exposure. PCT in our patient was associated with the risk factors of UROD mutation, HFE C282Y homozygosity, ferritin toxicity, and HCV. Biweekly phlebotomy of 450ml quickly decreased the patient’s ferritin levels and reduced iron overload toxicity. Finally, all patients with diagnosed PCT should be screened for hepatocellular carcinoma.
References
- Kowdley KV. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. 2004; 127: S79-86.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol. 2006; 20: 1201-1206.
- Sampietro M, Piperno A, Lupica L, Arosio C, Vergani A, Corbeta N, Malosio IDA, et al.. High prevalence of the His63Asp HFE mutation in italian patients with porphyria cutanea tarda. Hepatology. 1998; 27: 181-184.
- Stuart KA, Busfield F, Jazwinska EC, Gibson P, Butterworth LA, Cooksley WG, et al. The C282Y mutation in the haemochromatosis gene (HFE) and hepatitis C virus infection are independent cofactors for porphyria cutanea tarda in australian patients. J Hepatol. 1998; 28: 404-409.
- Powell LW. Diagnosis of hemochromatosis. Semin Gastrointest Dis. 2002; 13: 80-88.
- Steinberg KK, Cogswell ME, Chang JC, Caudill SP, McQuillan GM, Bowman BA, et al. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the united states. JAMA. 2001; 285: 2216-2222.
- Ajioka RS, Phillips JD, Weiss RB, Dunn DM, Maria WS, Proll SC, Katze MG, et al. Down-regulation of hepcidin in porphyria cutanea tarda. Blood. 2008; 112: 4723-4728.
- Christiansen AL, Aagaard L, Krag A, Rasmussen LM, Bygum A. Cutaneous porphyrias: Causes, symptoms, treatments and the danish incidence 1989- 2013. Acta Derm Venereol. 2016; 96: 868-872.
- Elder GH, Roberts AG, de Salamanca RE. Genetics and pathogenesis of human uroporphyrinogen decarboxylase defects. Clin Biochem. 1989; 22: 163-168.
- Herrero C, Ozalla D, Sala M, Otera R, Silva MS, Lecha M, et al. Urinary porphyrin excretion in a human population highly exposed to hexachlorobenzene. Arch Dermatol. 1999; 135: 400-404.
- Fung MA, Murphy MJ, Hoss DM, Berke A, Grant-Kels JM. The sensitivity and specificity of “caterpillar bodies” in the differential diagnosis of subepidermal blistering disorders. Am J Dermatopathol. 2003; 25: 287-290.
- Kim KH, Oh KY. Clinical applications of therapeutic phlebotomy. J Blood Med. 2016; 7: 139-144.
- Pandya AG, Nezafati KA, Ashe-Randolph M, Yalamanchili R. Deferasirox for porphyria cutanea tarda: A pilot study. Arch Dermatol. 2012; 148: 898-901.
- Anderson KE, Goeger DE, Carson RW, Lee SM, Stead RB. Erythropoietin for the treatment of porphyria cutanea tarda in a patient on long-term hemodialysis. N Engl J Med. 1990; 322: 315-317.
- Mogl MT, Pascher A, Presser SJ, Schwabe M, Neuhaus P, Nuessler NC. An unhappy triad: Hemochromatosis, porphyria cutanea tarda and hepatocellular carcinoma-a case report. World J Gastroenterol. 2007; 13: 1998-2001.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, Obando J, Di Bisceqlie A, Tattrie C, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in north america. Hepatology. 1998; 27: 1661-1669.
- Fernandez I, Castellano G, de Salamanca RE, Colina F, de la camara AG, Moran MJ, et al. Porphyria cutanea tarda as a predictor of poor response to interferon alfa therapy in chronic hepatitis C. Scand J Gastroenterol. 2003; 38: 314-319.